
Дилатационная кардиомиопатия (ДКМП) — заболевание миокарда, характеризующееся прогрессирующим увеличением размеров камер сердца и снижением сократительной способности миокарда; оно является наиболее частой причиной тяжелой сердечной недостаточности, при которой требуется трансплантация сердца [1]. Частота развития заболевания в популяции составляет около 36,5:100 000 населения, однако даже эти данные могут быть заниженными [2].
В 20—30% случаев ДКМП имеет семейный характер [3]. Заболевание генетически гетерогенное, в настоящее время известны более 100 генов, мутации в которых могут быть причиной кардиомиопатии (КМП) [4]. Поэтому ДНК-диагностика этого заболевания остается исключительно сложной задачей, а данные о клинических особенностях различных генетических форм заболевания весьма ограничены. В 2004 г. впервые показана роль мутаций в гене SCN5A как причина ДКМП, сопровождающейся нарушениями ритма и проводимости [5]. В международном каталоге наследственных заболеваний OMIM этот генетический вариант КМП обозначен ДКМП, тип 1Е, номер заболевания MIM:*601154 [6]. Этот ген кодирует α-субъединицу натриевого канала Nav1.5, ответственного за основной деполяризующий компонент потенциала действия кардиомиоцитов (КМЦ), входящий натриевый ток INa [7]. Натриевый входящий деполяризующий ток служит пусковым механизмом развития потенциала действия в КМЦ, результирующий деполяризующий ток — сигналом, инициирующим сокращение саркомерных белков отдельного КМЦ. Благодаря системе межклеточных контактов КМЦ и клеток проводящей системы сердца эти периодические деполяризации лежат в основе синхронного и ритмичного сокращения камер сердца [7].
Мутации в гене SCN5A являются причиной различных нарушений ритма сердца, таких как синдром удлиненного интервала QT, синдром Бругада, синдром слабости синусного узла, прогрессирующего нарушения проводимости [7].
Частота опосредованных SCN5A случаев в общей группе больных с ДКМП относительно невелика — около 3% [8]. Однако среди больных, у которых ДКМП сочетается с прогрессирующим поражением проводящей системы и наджелудочковыми и/или желудочковыми нарушениями ритма, их доля значительно выше. Согласно рекомендациям HRS/ESHRA (Heart Rhythm Assocoation/European Society of Heart Rhythm), больным с сочетанием предположительно первичной ДКМП и нарушениями проводимости (атриовентрикулярная — АВ-блокада любой степени) расширенное или выборочное (гены SCN5A и LMNA) генетическое исследование является рекомендованным [9].
Материал и методы
Проведено клиническое и генетическое обследование членов семьи DCM_83. Клинико-генеалогический анализ заключался в сборе и анализе семейных данных, в том числе архивного материала за период с 1992 по 2013 гг. Клиническое и инструментальное обследование включало детальный осмотр, электрокардиографию, суточное холтеровское мониторирование, ультразвуковое исследование (УЗИ) сердца и брюшной полости, мультиспиральную компьютерную томографию сердца (МСКТ), оценку функции внешнего дыхания. Лабораторные исследования включали общий и биохимический анализ крови, коагулограмму, исследование уровня гормонов щитовидной железы, оценку функции почек, ПЦР кардиотропных вирусов в крови. Получено информированное согласие от членов семьи на генетическое исследование, которое включало прямое секвенирование по Сенгеру кодирующей последовательности и прилегающих интронных областей гена SCN5A у пробанда, а также секвенирование экзона 10 (направленный поиск мутации) у членов семьи.
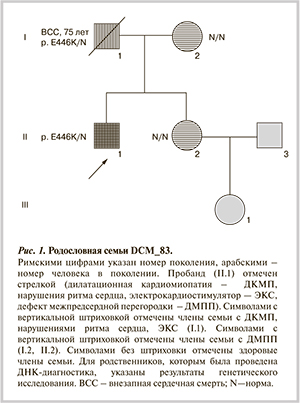 Клиническое наблюдение. В отделение хирургического лечения дисфункции миокарда ФГБУ РНЦХ им. акад. Б.В. Петровского РАМН обратился пациент 52 лет за обследованием и уточнением тактики лечения. Диагноз при поступлении: ДКМП, имплантация электрокардиостимулятора (ЭКС) в 2007 г. (в возрасте 46 лет), оперированный в 1992 г. врожденный порок сердца (дефект межпредсердной перегородки — ДМПП).
Клиническое наблюдение. В отделение хирургического лечения дисфункции миокарда ФГБУ РНЦХ им. акад. Б.В. Петровского РАМН обратился пациент 52 лет за обследованием и уточнением тактики лечения. Диагноз при поступлении: ДКМП, имплантация электрокардиостимулятора (ЭКС) в 2007 г. (в возрасте 46 лет), оперированный в 1992 г. врожденный порок сердца (дефект межпредсердной перегородки — ДМПП).
Семейный анамнез пациента был отягощен по заболеваниям сердца (рис. 1). Отец длительно наблюдался с диагнозом ДКМП, расширение всех камер сердца, синдром Фредерика, по поводу которого был неоднократно реимплантирован ЭКС, умер внезапно в возрасте 75 лет. У матери и сестры диагностирован ДМПП, по поводу чего сестра также оперирована в молодом возрасте. При обследовании 65-летней сестры больного не выявлено признаков снижения локальной или глобальной сократимости миокарда, расширения полостей сердца. При генетическом исследовании у нее также не выявлено изменений в гене SCN5A. Таким образом, врожденный порок сердца (ДМПП), диагностированный у 3 членов семьи, является самостоятельным наследственным заболеванием, наследующимся независимо от КМП, и не является частью фенотипического проявления мутации p.E446K в гене SCN5A.
Из анамнеза обследованного больного: в росте и развитии от ровесников не отставал, физические нагрузки переносил удовлетворительно. В 1992 г. впервые выполнили УЗИ сердца, по данным которого выявлен ДМПП. В том же году выполнена пластика ДМПП заплатой из ксеноперикарда. В амбулаторной карте имеется информация о перенесенном в 1994 г. инфекционном эндокардите на фоне ревматического порока аортального клапана с преобладанием недостаточности.
В возрасте 40 лет впервые зарегистрирована полная блокада левой ножки пучка Гиса (БЛНПГ). В 42 года нарушения ритма были более выраженными: БЛНПГ, частые пароксизмы желудочковой тахикардии; выполнена коронарография, при которой стенотическое поражение коронарных артерий не выявлено. Пациент постоянно принимал β-адреноблокаторы. В возрасте 43 лет (2004 г.) диагностированы расширение камер сердца, снижение сократительной способности миокарда, впервые поставлен диагноз первичной ДКМП.
В возрасте 46 лет (2007 г.) по данным УЗИ сердца: конечный диастолический размер (КДР) левого желудочка (ЛЖ) 62 мм, конечный систолический (КСР) размер ЛЖ 45 мм, правый желудочек (ПЖ) 38 мм, левое предсердие (ЛП) 70 мм, межжелудочковая перегородка (МЖП) 13 мм, фракция выброса (ФВ) ЛЖ 52%, снижение сократительной способности миокарда. Нарушения ритма: постоянная форма фибрилляции предсердий, брадисистолия, АВ-блокада III степени, желудочковая диссинхрония (межжелудочковая задержка до 90 мс). Имплантирован кардиостимулятор Аxiоs SR, Polyrox 60BP, Corox OTW 75 UP в коронарный синус для бивентрикулярной стимуляции. При выписке пациенту назначены ацетилсалициловая кислота 150 мг/сут, эналаприл по 2,5 мг 2 раза в сутки. В этом же году пациенту проведено генетическое исследование и выявлена мутация p.E446K в гене SCN5A. Таким образом, диагноз первичной ДКМП, тип 1Е (MIM:*601154) молекулярно-генетическими методами был подтвержден (рис. 2, см. на цв. вклейке).
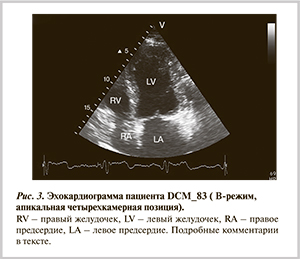
В 2013 г. пациент был приглашен на плановую госпитализацию с целью катамнестического наблюдения (6 лет), проверки функции стимулятора и уточнения тактики лечения. На момент госпитализации основные жалобы связаны с одышкой при подъеме по лестнице до 3-го этажа, перебои в работе сердца, периодические боли в левой половине грудной клетки без связи с физической нагрузкой. При обследовании: частота сердечных сокращений 69 уд/мин, ритм кардиостимулятора. Данные эхокардиографии сердца (рис. 3): увеличение размеров ЛЖ, КДР ЛЖ 6,7 см, КСР ЛЖ 5,0 см, МЖП 1,5 см, задняя стенка (ЗС) ЛЖ 1,1 см, боковая стенка ЛЖ 1,5 см, конечный диастолический объем 175 мл, конечный систолический объем 78 мл, ударный объем 97 мл, ФВ ЛЖ 55% (по Симпсону). Верхушка закруглена, повышенная трабекулярность в области верхушки, в полости ЛЖ лоцируется несколько диагональных хорд, аномально прикрепленная папиллярная мышца в области верхушечных сегментов боковой стенки; асинхронное сокращение верхушечных сегментов боковой и передней стенок. Увеличение размеров ПЖ (до 3,4 см), ЛП (6,1×7,5 см), правого предсердия (3,9×5,4 см). Левая атриомегалия. Комбинированный аортальный порок (аортальная недостаточность II степени, незначительный стеноз аортального клапана). Расширение восходящего отдела аорты, преимущественно на уровне синусов. Незначительное снижение локальной систолической функции гипертрофированного ЛЖ, глобальная систолическая функция его сохранена. Межжелудочковая (лево-правая) задержка: –10 мс (норма <40 мс).
Для уточнения причины изолированной левой атриомегалии, не укладывающейся в рамки последствий корректированного более 20 лет назад врожденного порока сердца (ДМПП), выполнена МСКТ сердца, по данным которой дополнительного источника гемодинамической перегрузки ЛП не выявлено.
Диагноз при выписке: ДКМП генетически детерминированная, тип 1Е (MIM:*601154). ВПС: ДМПП, пластика ДМПП в 1992 г. заплатой из ксеноперикарда. Нарушения ритма и проводимости сердца: полная БЛНПГ, желудочковая экстрасистолия, класс по Лауну 4Б, «пробежки» ЖТ? до 7 комплексов (класс по Lown—Wolff—Ryan V), фибрилляции предсердий, перманентная форма, брадисистолия. Имплантация ЭКС Аxios SR в августе 2007 г., реимплантация ЭКС Axios DR в октябре 2007 г., бивентрикулярная стимуляция. Хроническая сердечная недостаточность III функционального класса по классификации NYHA. Недостаточность кровообращения 2А степени. Комбинированный аортальный порок сердца: умеренный стеноз, недостаточность II степени. Эктазия восходящей аорты.
При выписке даны следующие рекомендации: регулярные проверки кардиостимулятора, регулярная эхокардиография для оценки состояния аортального клапана, при прогрессировании — протезирование клапана; эналаприл 2,5 мг 2 раза в сутки; эплеренон (по 25 мг ежедневно) (контроль уровня креатинина, уровня калия крови 1 раз в месяц); соталол 40 мг 2 раза в сутки; ацетилсалициловая кислота 150 мг 1 раз в сутки; порошок калия хлорида 1000 мг (под контролем уровня калия крови 1 раз в месяц); магнерот 1 таблетка 3 раза в сутки ежедневно; омепразол 20 мг по 1 капсуле в течение 1 мес после выписки.
Обсуждение
Представленный клинический случай является первым в России длительным наблюдением генетически верифицированной КМП, тип 1Е (MIM: *601154).
Спектр проявлений в целом типичен для этой формы заболевания: дилатация камер, фибрилляция предсердий, прогрессирующее нарушение АВ-проведения, желудочковые нарушения ритма сердца. При анализе опубликованных клинических особенностей больных ДКМП с мутациями в гене SCN5A частота наджелудочковых нарушений ритма составляла 86%, фибрилляция предсердий — 60%, нарушения проводящей системы — 60% [10]. Частым признаком этой формы КМП также является прогрессирующая дилатация предсердий без явных предрасполагающих гемодинамических факторов (предсердная КМП?) [9], что мы также наблюдали у нашего пациента.
Как правило, генетически детерминированные формы ДКМП характеризуются неуклонно прогрессирующим течением и неблагоприятным прогнозом, ограниченной эффективностью кардиотропной и антиаритмической терапии [11]. Поэтому стабильность сократительной способности и ФВ ЛЖ за истекшие 6 лет (53—55% за период с 2007 по 2013 г.) выделяют этот клинический случай из ряда других наследственных форм ДКМП, которые наблюдались в нашем центре.
Мутация p.E446K в гене SCN5A ранее описана как причина ДКМП с нарушениями ритма и проводимости у пациента с манифестацией заболевания в возрасте 24 лет [10]. Следует отметить, что в течение 3 лет наблюдения у пациента также имелось увеличение ФВ ЛЖ с 24 до 40% [11]. Еще один случай восстановления сократительной способности миокарда описан у пациента с ДКМП, множественными полиморфными желудочковыми экстрасистолами (ЖЭ) и мутацией p.R222Q в гене SCN5A [12]. Данная мутация приводит к появлению позднего входящего натриевого тока (увеличение суммарного натриевого тока), и как следствие, к постоянному смещению мембранного потенциала в сторону деполяризации [12]. При лечении больных родственников — носителей этой мутации, стандартная терапия, применяемая при сердечной недостаточности, была практически неэффективна. Однако после того как был поставлен генетический диагноз и получены данные о функциональных свойствах мутации, больным были назначены препараты, блокирующие натриевые каналы, что привело к резкому снижению числа ЖЭ и восстановлению сократительных свойств миокарда ЛЖ через 6 мес от начала приема препаратов [12]. К сожалению, данных о биофизических характеристиках ионного канала Nav1.5, содержащего мутацию p.E446K, в настоящее время нет, что не позволяет предложить аналогичную тактику лечения для нашего пациента.
 Относительно медленное прогрессирование ДКМП [11], эффективность ресинхронизирующей терапии, а также имеющиеся описания восстановления сократительной способности миокарда при оптимальной коррекции нарушений ритма [13] свидетельствуют в пользу аритмогенного характера ремоделирования миокарда у больных c ДКМП 1Е. Нарушение функции натриевых каналов, неодинаковый уровень их экспрессии в различных отделах сердца могут вести к прогрессирующей электрической гетерогенности и диссинхронии при сокращении [14]. В свою очередь, желудочковая и предсердная хроническая аритмия/диссинхрония могут являться движущим механизмом аритмогенного ремоделирования. Поэтому ресинхронизирующая терапия представляется потенциально высокоэффективным подходом к лечению таких больных.
Относительно медленное прогрессирование ДКМП [11], эффективность ресинхронизирующей терапии, а также имеющиеся описания восстановления сократительной способности миокарда при оптимальной коррекции нарушений ритма [13] свидетельствуют в пользу аритмогенного характера ремоделирования миокарда у больных c ДКМП 1Е. Нарушение функции натриевых каналов, неодинаковый уровень их экспрессии в различных отделах сердца могут вести к прогрессирующей электрической гетерогенности и диссинхронии при сокращении [14]. В свою очередь, желудочковая и предсердная хроническая аритмия/диссинхрония могут являться движущим механизмом аритмогенного ремоделирования. Поэтому ресинхронизирующая терапия представляется потенциально высокоэффективным подходом к лечению таких больных.
В настоящее время описано уже более 10 мутаций в гене SCN5A, приводящих к развитию ДКМП в сочетании с широким спектром нарушений ритма сердца и проводимости [5, 8, 11, 13]. Спектр биофизических нарушений функций натриевого канала при этих мутациях также варьирует. Описаны как генетические варианты, приводящие к суммарному увеличению натриевого тока через измененный канал, так и мутации, приводящие к снижению проводимости натрия. Одна из замен, приводящих к развитию КМП, p.R219H, приводит к появлению стабильного входящего протонного тока «утечки» через мутантный ионный канал, что приводит к снижению рН и «закислению» внутриклеточной среды КМЦ [13]. Можно предположить, что длительно существующее нарушение рН внутри клетки может запускать механизмы метаболических расстройств и нарушения ионного баланса, подобные внутриклеточным нарушениям в условиях хронической ишемии.
Несмотря на достаточно подробную биофизическую характеристику мутантных вариантов белка Nav1.5, все этапы молекулярного патогенеза, ведущего от нарушения проводимости натрия к поражению проводящей системы и ремоделированию миокарда, окончательно неясны. Однако детальное понимание всех этапов (молекулярного, клеточного, органного) ремоделирования необходимо для выбора максимально эффективной персонализированной стратегии лечения на основе существующих методов или разработки новых подходов к терапии нарушений ритма и сердечной недостаточности.


