В практике врачей различных специальностей нередко встречаются заболевания, сопровождающиеся поражением различных органов и систем, в том числе и кожи. При этом бывает достаточно сложно быстро поставить правильный диагноз. Это связано с тем, что кожные высыпания недостаточно хорошо идентифицируются интернистами, а дерматологи сильно ограничены лабораторной и инструментальной диагностической базой.
В связи с этим мы уверены, что напоминание о редких синдромах, заболеваниях, реакциях будет полезным терапевтам, кардиологам, пульмонологам, нефрологам и врачам других специальностей.
GRAFT-VERSUS-HOST DISEASE
Graft-versus-host disease представляет собой реакцию «трансплантат против хозяина» (РТПХ) и является одним из наиболее тяжелых осложнений аллогенной трансплантации гемопоэтических стволовых клеток. Она возникает более чем у половины пациентов, подвергшихся такому вмешательству, и продолжает оставаться одной из главных причин смертности в посттрансплантационном периоде [1]. Частота развития острой и хронической РТПХ при трансплантации гемопоэтических стволовых клеток превышает таковую при трансплантации костного мозга: 67 против 54% [2].
РТПХ, развившаяся в первые 100 дней после трансплантации, является острой и характеризуется поражением кожи и ЖКТ (гепатит, энтерит). При хронической форме (100 дней после трансплантации) могут развиться системные симптомы с поражением кожи, ЖКТ, печени, легких и др. [3].
Поражение кожи может быть генерализованным или локализованным и проявляться разнообразной симптоматикой. Так, встречаются высыпания, сходные с красным плоским лишаем. При этом папулы имеют фиолетовую окраску, напоминая элементы красного плоского лишая, сопровождаются зудом, часто располагаются на ладонях и подошвах, но могут быть распространенными и иметь тенденцию к слиянию. После разрешения обычно остается интенсивная гиперпигментация с неправильными очертаниями (рис. 1).
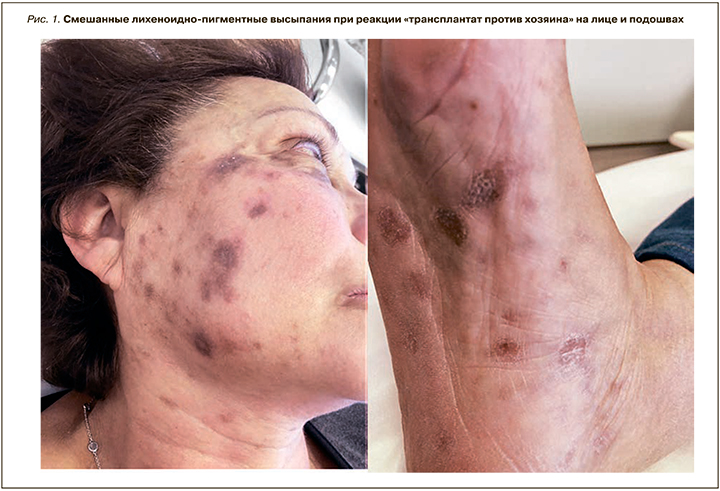
Знание кожных проявлений РТПХ позволяет направить пациента в гематологическое учреждение без траты времени на симптоматическое лечение. Рано начатое лечение РТПХ позволяет снизить летальность [4].
СИНДРОМ ЛУИ–БАР
Синдром Луи–Бар (атаксия–телеангиэктазия) относится к так называемым факоматозам – генетически обусловленным сочетанным поражениям кожи и нервной системы. В основе патологических изменений, сопровождающих этот синдром, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи–Бар относится к аутосомно-рецессивным заболеваниям, т.е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей. Он одинаково часто встречается у мальчиков и девочек, частота заболевания составляет 1 случай на 90 тыс. новорожденных [5, 6].
Наиболее часто синдром Луи–Бар начинает проявляться в возрасте от 5 мес до 3 лет. При этом отмечается появление мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание, качание туловища и головы во время движения. Характерна мозжечковая дизартрия, характеризующаяся невнятной скандированной речью. Также отмечается мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие. У пациентов имеется склонность к инфекционным заболеваниям – частым ОРВИ, пневмониям, имеющим затяжное течение и плохо поддающимся лечению. Нередки опухоли яичников, желудка, кожи [7].
Телеангиэктазии появляются в возрасте от 3 до 6 лет и изначально на конъюнктиве глазного яблока, затем на коже век, носа, лица и шеи, локтевых и коленных сгибах, предплечьях, тыльной поверхности стоп и кистей. Они могут также наблюдаться на слизистой оболочке мягкого и твердого нёба, но наиболее выражены в местах кожного покрова, где он подвергается воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». При этом кожа теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии [8].
Из других кожных проявлений встречаются веснушки, пятна цвета кофе с молоком, участки обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи–Бар схожими с клиникой пойкилодермии. У многих больных наблюдаются сухость кожи и участки гиперкератоза. Может наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне, проявления псориаза. Мы наблюдали редкий случай развития у девочки 5 лет липоидного некробиоза (рис. 2) [9].
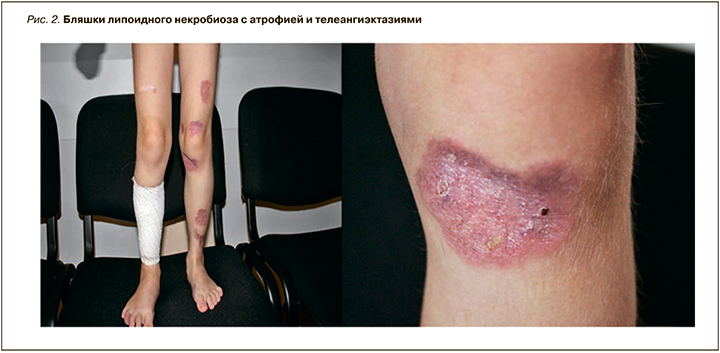
Из инструментальных способов диагностики синдрома Луи–Бар могут применяться ультразвуковое исследование (УЗИ) тимуса, магнитно-резонансная томография (МРТ) головного мозга, фарингоскопия, риноскопия, рентгенография легких. При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений [10, 11].
Синдром Луи–Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю–Ослера, атаксией Пьера–Мари, синдромом Гиппеля–Линдау и др.
Диагностика атаксии–телеангиэктазии требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи–Бар должен пройти обследование не только у невролога, но и у отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога, дерматолога.
СИНДРОМ LEOPARD
Синдром LEOPARD включает комплекс дисморфогенетических расстройств:
- лентиго (L);
- электрокардиографические нарушения проводимости (E);
- глазной гипертелоризм (O);
- стеноз легочной артерии (P);
- аномалии половых органов (A);
- замедление роста (R);
- глухоту (D).
Молекулярные исследования показали, что синдром LEOPARD – результат мутации в гене PTPN11 (90%) или RAF1 (10%). Следствием этого становится нарушение функции фермента протеин-тирозин-фосфатазы SHP-2, что, в свою очередь, ведет к изменению пролиферации клеток, нарушению их дифференцировки и потери жизнеспособности. Тип наследования аутосомно-доминантный. 70% случаев заболевания являются семейными. Синдром встречается с частотой менее 1:1 000 000 [12, 13].
Кожные высыпания представлены полигональными или неправильной формы пигментными пятнами темно-коричневого, черного цвета, размером от 2–5 мм до 1–1,5 см в диаметре. Пятна присутствуют у 90% пациентов на лице, шее и верхней части туловища, на ладонях, ступнях. Могут они появляться и на склерах. Слизистые оболочки при этом не поражаются. При тщательном обследовании кожи можно обнаружить другие кожные нарушения, например подмышечные веснушки, локализованные гипопигментации, ониходистрофии (рис. 3) [14].
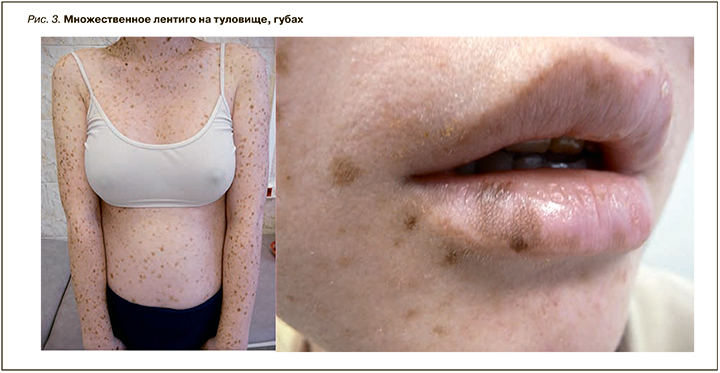
У 25% пациентов имеется характерное лицо с широко расставленными глазами (гипертелоризм), у 35% выявляются различные черепно-лицевые и скелетные аномалии. У 30% больных наблюдается умственная отсталость, обычно легкой степени, около 25% страдают нейросенсорной потерей слуха. У трети пациентов отмечается невысокий рост, который становится очевидным вскоре после рождения. У части больных можно выявить недоразвитие половых органов, гипоспадию и др. [15].
Несмотря на частое развитие пороков сердца, большинство пациентов с синдромом LEOPARD остаются бессимптомными.
Другие признаки заболевания включают стоматологические аномалии, прогнатизм, низко посаженные уши, птоз, «готическое» нёбо и др. [16].
Диагностика синдрома LEOPARD должна включать следующие исследования: компьютерную томографию (КТ) или МРТ головы, рентгенографию скелета, эхокардиографию, обследование мочеполовой системы, УЗИ, электрокардиографию (ЭКГ).
Прогноз заболевания в основном определяется выраженностью сердечно-сосудистых осложнений. Большинство пациентов с синдромом LEOPARD могут вести нормальную жизнь.
ПСЕВДОСАРКОМА КАПОШИ
Псевдосаркома Капоши – редкое поражение кожи, преимущественно нижних конечностей, клинически схожее с саркомой Капоши. Причиной этой патологии служит порок эмбрионального развития сосудистой системы. В связи с этим различают два варианта псевдосаркомы:
- ангиодермит, вызванный хронической венозной недостаточностью и описанный в 1965 г. Mali J.W. et al.;
- поражение кожи, связанное с врожденной артериовенозной недостаточностью с множественными артериовенозными фистулами и описанное в 1967 г. Bluefarb S.M. и Stewart W.M. [17].
Псевдосаркома Капоши типа Mali наблюдается у мужчин в возрасте 40–50 лет. При этом на фоне хронической венозной недостаточности на коже голеней и стоп появляются пятна и крупные бляшки синюшно-коричневого цвета размером до 10 см и более. Бляшки сопровождаются болезненностью и часто изъязвляются. Нередко в области лодыжек развиваются сетчатая пигментация, очаги атрофии (рис. 4) [18].
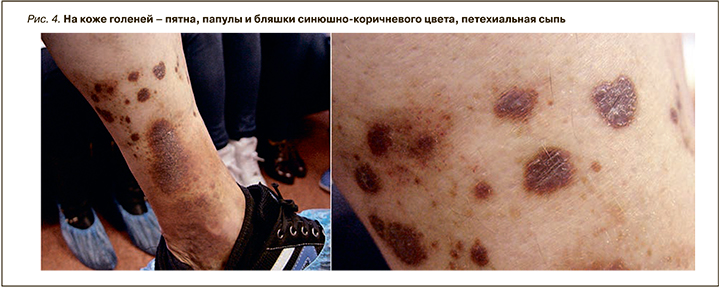
При втором типе псевдосаркомы (Bluefarb и Stewart) клиническая картина схожа с болезнью Капоши, однако кожные и сосудистые проявления носят односторонний характер. Заболевание проявляется в возрасте 20–30 лет на нижней половине одной из голеней, в области щиколотки и на тыле I, II, III пальцев стопы в виде пятен, папул и бляшек с четкими границами, синюшно-коричневого и ливидного цвета. Возможны изъязвления и папилломатозные разрастания. Часто отмечается отек и даже удлинение всей конечности, варикозное расширение вен. Эти симптомы сопровождаются резкой болезненностью. При артерио- и флебографии выявляется ангиодисплазия с множественными артериовенозными фистулами [19].
Дифференциальный диагноз проводят в первую очередь с саркомой Капоши, а также ангиокератомой, красным плоским лишаем. Интересно, что гистопатологические изменения при обеих формах схожи с саркомой Капоши. Для подтверждения формы Bluefarb и Stewart необходима артериография.
Терапию при псевдосаркоме Капоши типа Mali проводят как при венозной недостаточности. При втором типе заболевания (Bluefarb и Stewart) возможно хирургическое вмешательство с ушиванием шунтов или даже ампутацией конечности.
ГРАНУЛЕМАТОЗ ВЕГЕНЕРА
Гранулематоз Вегенера развивается как аутоиммунное гранулематозное воспаление стенок сосудов, с вовлечением верхних дыхательных путей, легких, глаз, почек, кожи и других органов. Он относится к системным антинейтрофильным цитоплазматическим антитело-ассоциированным некротизирующим васкулитам. Заболевание может начаться в любом возрасте (в среднем около 40 лет), несколько чаще у мужчин, хотя дети болеют редко. Около 15% больных с этим диагнозом моложе 19 лет [20].
Этиология гранулематоза Вегенера неизвестна, возможно, определенную роль играет хроническая очаговая инфекция (носоглоточная). Имеет значение и гиперреактивность гуморального звена иммунитета: у больных наблюдается повышение сывороточных и секреторных IgA, G, E, выявляются циркулирующие иммунные комплексы (ЦИК), IgG-аутоантитела. Заболевание связано с наличием антигенов гистосовместимости HLA В7, В8 и DR2, что говорит об определенной генетической предрасположенности к нему. У большинства больных имеются антитела к цитоплазме нейтрофилов, преимущественно к протеазе-3 [21].
Гранулематоз Вегенера развивается постепенно: поражение верхних дыхательных путей встречается у 92% пациентов и проявляется ринитом с язвенно-некротическими изменениями слизистой оболочки придаточных пазух, гортани, трахеи. Возможно появление гнойного отита.
Поражение легких наблюдается у 85–90% больных и проявляется кашлем, одышкой, кровохарканьем и болями в груди. У 1/3 больных рентгенологические признаки могут не сопровождаться клиническими проявлениями легочной патологии.
Поражение глаз, которое встречается в 52% случаев, проявляется в виде конъюнктивита, дакриоцистита, эписклерита, склерита, гранулематозного склероувеита, иридоциклита, гранулемы ретробульбарной клетчатки и экзофтальма.
Поражение сердца выявляется в 8% случаев и ведет к перикардиту, коронарному васкулиту, инфаркту миокарда, поражению митрального и аортального клапанов, атриовентрикулярной блокаде.
Поражение нервной системы отмечается у 23% больных и включает нейропатии черепных нервов, множественную мононейропатию, изредка церебральный васкулит и гранулемы головного мозга.
Поражение почек имеет место у 77% больных и преобладает в клинической картине. Оно может ограничиваться легким гломерулонефритом с протеинурией, гематурией и эритроцитарными цилиндрами, но при почечной недостаточности быстро прогрессирует.
Во время обострений гранулематоза Вегенера появляются неспецифические симптомы – недомогание, слабость, артралгия, снижение аппетита, похудание, лихорадка [22].
Поражение кожи отмечается у 46% больных гранулематозом Вегенера в виде папул, везикул, пальпируемой пурпуры. Однако типичными проявлениями считаются узлы и язвы, обусловленные некротическим ангиитом дермальных сосудов с их тромбозом и некрозом.
Поражение слизистой оболочки полости рта встречается почти у всех больных и проявляется гранулематозными разрастаниями в области десен, нёба, дужек, миндалин. Они имеют бугристую поверхность, застойно-красный цвет, плотную консистенцию и быстро распадаются с образованием язв разной глубины. У ряда больных процесс сопровождается увеличением подчелюстных лимфоузлов. Может произойти некроз тканей пародонта, прободение твердого нёба, разрушение мягких тканей и костей средней трети лица. При этом при осмотре больного ощущается зловонный запах (рис. 5) [23, 24].
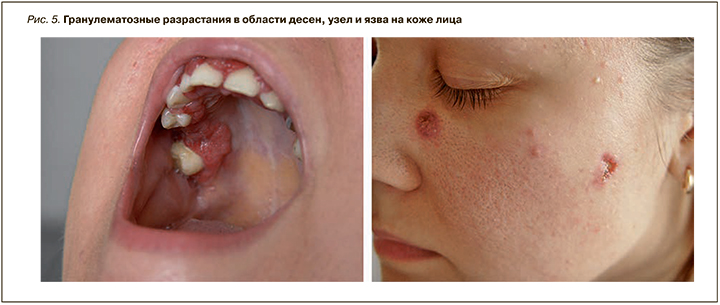
Выделяют две формы гранулематоза Вегенера – локализованную и генерализованную. Первая начинается с поражения верхних дыхательных путей или глаз. Реже первично поражается слизистая оболочка рта и глотки. При генерализованной форме процесс начинается с поражения трахеобронхиального дерева и легких и клинически выражается лихорадкой различной выраженности, полиморфными высыпаниями, кашлем с гнойно-кровянистой мокротой. Затем присоединяются симптомы поражения других органов. Возможны артриты, артралгии и миалгии, анемия, нейтрофильный лейкоцитоз, ускорение скорости оседания эритроцитов (СОЭ). Может развиться хондрит ушных раковин и др.
Прогноз гранулематоза Вегенера неблагоприятный. Без своевременно начатого лечения смертельный исход может наступить в течение 6–12 мес. Смерть наступает чаще от почечной или сердечно-сосудистой недостаточности [25].
Если говорить о диагностике гранулематоза Вегенера, то у 25% больных в начальной стадии нет признаков поражения почек или легких. Только у 50% пациентов болезнь диагностируется в первые 3–6 мес от ее начала, а у 7% это не диагностируется даже в течение 5–16 лет от появления первых клинических симптомов.
Характерными лабораторными находками при гранулематозе Вегенера выступают значительное повышение СОЭ, анемия, лейкоцитоз, гипергаммаглобулинемия (в основном за счет IgA), появление ревматоидного фактора, мочевой синдром, свойственный гломерулонефриту. Антитела к протеазе-3 (АНЦА) находят у 90% больных с поражением дыхательных путей и почек и лишь у 70% больных без поражения почек.
Гистологический диагноз характеризуется обнаружением в биоптате некротического васкулита, сопровождающегося гранулематозным воспалением [26].
Дифференциальный диагноз проводится со срединной гранулемой лица, лимфоматоидным гранулематозом, синдромами Черджа–Стросс, Гудпасчера, опухолями верхних дыхательных путей и легких, кожно-слизистым лейшманиозом, склеромой, другими инфекционными и неинфекционными гранулематозами.
