
Гипертрофическая кардиомиопатия (ГКМП) — генетически обусловленное заболевание, которое может иметь как благоприятное, так и тяжелое течение, характеризующееся развитием фатальных осложнений, приводящих к инвалидизации и смерти людей молодого возраста [1]. Эпидемиология ГКМП в России практически не изучена, что обусловлено, с одной стороны, отсутствием крупных популяционных исследований, а с другой, — часто бессимптомным течением, при котором единственным проявлением заболевания может быть внезапная сердечная смерть (ВСС). По расчетным данным, составленным с учетом коэффициентов в ходе эпидемиологических исследований стран Европы и США, возможная частота ВСС в России составляет 141—460 тыс. в год. [2]
Ранняя оценка прогноза течения заболевания, своевременное начало и правильный выбор тактики лечения существенно снижают уровень сердечно-сосудистой инвалидизации и смертности у этих больных. Поэтому актуальной проблемой остается поиск методов ранней диагностики ГКМП и способов оценки риска развития неблагоприятных клинических исходов.
В настоящее время продолжаются активный поиск и исследование многочисленных потенциальных генов-модификаторов, способных влиять на фенотипические проявления различных сердечно-сосудистых заболеваний (ССЗ), в частности ГКМП.
Доказано, что ренин-ангиотензин-альдостероновая система (РААС) участвует в развитии ремоделирования сердечно-сосудистой системы при ГКМП (развитие гипертрофии миокарда, пролиферация фибробластов, нарушение фибринолиза, стимуляция синтеза коллагена, накопление внеклеточного матрикса), что может иметь большое значение для формирования вариантов клинического течения [3—5].
Актуальной представляется оценка маркеров нейрогуморальных систем у больных ГКМП с различными вариантами течения, так как эта область остается мало изученной.
Целью нашего исследования послужило изучение роли полиморфизмов генов, кодирующих компоненты РААС (AGT, AGTR1, CYP11B2, CMA1) и маркеров, характеризующих эту систему (АСЕ и АТII), у больных ГКМП с различными вариантами клинического течения.
Материал и методы
 Обследованы 58 больных ГКМП (24 мужчины и 34 женщины), наблюдающихся в клинике госпитальной терапии №1 Первого МГМУ им. И.М. Сеченова, не связанных узами родства, средний возраст которых составил 48,4±13,6 года. В контрольную группу вошли 54 здоровых добровольца (22 мужчины, 32 женщины), средний возраст которых составил 49,3±13,5 года и не отличался от такового у пациентов исследуемой группы (р=0,95).
Обследованы 58 больных ГКМП (24 мужчины и 34 женщины), наблюдающихся в клинике госпитальной терапии №1 Первого МГМУ им. И.М. Сеченова, не связанных узами родства, средний возраст которых составил 48,4±13,6 года. В контрольную группу вошли 54 здоровых добровольца (22 мужчины, 32 женщины), средний возраст которых составил 49,3±13,5 года и не отличался от такового у пациентов исследуемой группы (р=0,95).
Диагноз ГКМП устанавливали согласно рекомендациям по диагностике и лечению больных ГКМП. В отсутствие фенотипических проявлений проводили молекулярно-генетическое исследование.
Больные были разделены на группы по вариантам клинического течения: стабильное течение (n=21, 36,2%); вариант с фибрилляцией предсердий — ФП (n=10, 17,2%); прогрессирующее течение ГКМП (n=27, 46,6%). Все пациенты проходили стандартное клиническое обследование.
Эхокардиографию (ЭхоКГ) проводили на аппарате Vivid7 Dimension/Vivid 7 PRO версия 6.0.х (Германия) по методике двухмерной ЭхоКГ с использованием М- и В-режимов, а также с использованием импульсно-волнового и непрерывно-волнового допплеровских режимов. Оценивали размеры камер сердца, основные показатели гипертрофии миокарда, в том числе толщину межжелудочковой перегородки (ТМЖП) и задней стенки левого желудочка (ТЗСЛЖ). Определяли также показатели диастолической дисфункции, нарушения глобальной и локальной сократимости миокарда. На основании полученных данных, согласно стандартизованным математическим формулам, производили вычисление следующих параметров: массы миокарда ЛЖ (ММЛЖ), индекса массы миокарда ЛЖ (ИММЛЖ), отношения между наполнением ЛЖ в диастолу – пик Е и систолу предсердий – пик А (Е/А), индекса объем-масса (ИОМ), предсердно-желудочкового отношения (ПЖО) [6].
Анализ полиморфизмов генов-модификаторов. У всех больных определяли полиморфизмы генов-модификаторов, характеризующих РААС: химазы (СМА1 A(-1903)G rs1800875), ангиотензиногена (AGТ M235T rs699), рецепторов ангиотензина II 1-го типа (AGTR1 A1166C rs5186), альдостеронсинтазы (CYP11B2 -344 T/C rs1799998). В работе использовали образцы ДНК, выделенные из периферической крови. Проводили полимеразную цепную реакцию (ПЦР)синтеза ДНК и рестрикционный анализ амплифицированных фрагментов ДНК. Амплификация выполнена с помощью ПЦР на термоциклере Терцик. Разделение амплифицированных фрагментов ДНК проводили при помощи электрофореза.
Определение биологических маркеров. В соответствии с поставленными задачами, 40 больным из группы ГКМП и 39 из группы контроля проводили определение биологических маркеров в сыворотке крови. На основании количественного твердофазного иммуноферментного анализа (ИФА) проводили определение ангиотензинпревращающего фермента (AПФ). Определение маркера ангиотензина II (АТII) выполняли на основе количественного твердофазного ИФА с использованием набора EA3501-1 BCM Diagnostics Ангиотензин II, 96.
Статистическую обработку полученных данных выполняли с использованием пакета прикладных программ MicrosoftExcel 2010, Statistica (V. 6.0). Вид распределения для количественных признаков определяли с помощью критерия Шапиро—Уилка. Для сравнения количественных признаков, распределенных ненормально, применяли U-критерий Манна—Уитни либо критерий Крускала—Уоллиса. При попарном сравнении частот генотипов аллелей использовали точный двусторонний критерий Фишера. Для 3 категорий и более применяли однофакторный дисперсионный анализа (ANOVA), где вероятность случайного различия определяли по критерию Фишера p(F). Критерий p(χ2) использовали для оценки признаков, измеряемых в номинальной (категорийной) шкале.
Результаты
 В ранее проведенных нами исследованиях [5], а также исследованиях других авторов [7] установлено, что ряд генотипов РААС ассоциирован с выраженной гипертрофией миокарда и более тяжелым течением ГКМП (генотип СС полиморфизма AGТ M235T; генотип AG СМА1 A(-1903)G; аллель С AGTR1 A1166C и генотип СС CYP11B2 -344 T/C). В связи с этим мы распределили всех больных на группы в зависимости от наличия или отсутствия у них указанных неблагоприятных генотипов.
В ранее проведенных нами исследованиях [5], а также исследованиях других авторов [7] установлено, что ряд генотипов РААС ассоциирован с выраженной гипертрофией миокарда и более тяжелым течением ГКМП (генотип СС полиморфизма AGТ M235T; генотип AG СМА1 A(-1903)G; аллель С AGTR1 A1166C и генотип СС CYP11B2 -344 T/C). В связи с этим мы распределили всех больных на группы в зависимости от наличия или отсутствия у них указанных неблагоприятных генотипов.
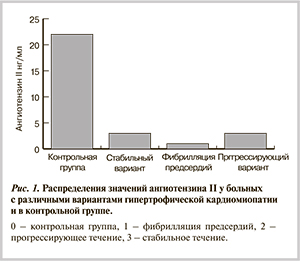
Маркеры нейрогуморальных систем. Выявлено значительное различие концентраций АТII между группой больных ГКМП и контрольной (критерий Фишера p(F)=0,000; критерий Крускала—Уоллиса p(H)=0,000; медианный тест p(χ²)=0,010). В группе ГКМП средний уровень колебался от 0,70±0,467 до 2,27±1,393 нг/мл, в то время как в контрольной группе средний уровень АТII составлял 33,36±8,226 нг/мл (рис. 1).
Анализ влияния полиморфизмов генов модификаторов на уровень маркера ATII у больных ГКМП не выявил статистически значимых различий. Однако прослеживалась тенденция к ассоциации с изучаемым полиморфизмом ангиотензиногена — AGТ M235T (р=0,073).
При анализе распределения уровней АТII в зависимости от аллельного полиморфизма AGТ M235T отмечено, что он был наиболее высоким у носителей генотипа TT (1,19±0,452 нг/мл), в то время как у носителей генотипов TC и CC средние уровни составили 0,597±0,301 и 0,158±0,404 нг/мл соответственно, однако эти различия лежат в пределах случайных колебаний.
Определялась прямая корреляция маркера АТII с ТЗСЛЖ (r=0,648; p=0,00001).
Отмечено повышение средних уровней маркера АПФ в группе прогрессирующего течения — 191,42±15,46 нг/мл. Однако это повышение оказалось статистически незначимым (критерий Фишера p(F)=0,78; критерий Крускала—Уоллиса p(H)=0,61). На содержание АПФ в крови оказывали значимое влияние изучаемые полиморфизмы гена альдостеронсинтазы (р=0,007) и в меньшей степени – гена химазы (р=0,072).
У носителей генотипа ТТ полиморфизма CYP11B2 -344 T/C обнаружено более высокое содержание АПФ (144,0—364,0 нг/мл) по сравнению с таковым у носителей аллеля С (104,0—219,5 нг/мл). Однако при этом следует учесть, что генотип TT найден всего у 3 больных с известным уровнем АПФ, тогда как в других группах имеется по 7 и 8 наблюдений. Для носителей генотипа СТ свойственно увеличение концентрации АПФ в крови по мере нарастания тяжести заболевания.
Отмечено снижение уровня АПФ у носителей генотипа GG СМА1 A(-1903)G в группе больных ГКМП с прогрессирующим вариантом течения (188,0±8,0 нг/мл) по сравнению с таковым у больных ГКМП с ФП (346,0 нг/мл).
В то же время для носителей генотипа АА характерно увеличение уровня АПФ при нарастании тяжести заболевания (от 122,0±18,5 до 218,0 нг/мл).
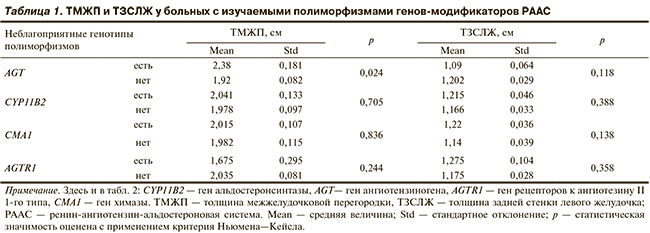
При анализе показателей гипертрофии миокарда выявлено, что для носителей неблагоприятного генотипа полиморфизма M235T гена ангиотензиногена характерны значимое увеличение ТМЖП (2,380±0,181 см), ММЛЖ (263,506±16,696 г) и тенденция к увеличению ТЗСЛЖ (1,09±0,064 см) и ИММЛЖ (138,527±11,276 г/м2).
Однофакторный дисперсионный анализ показал также наличие тенденции к росту ТЗСЛЖ у носителей генотипа AG изучаемого полиморфизма гена химазы (1,22±0,036 см; табл. 1, 2).
Анализ взаимосвязи степени стенокардии с наличием неблагоприятных генотипов изучаемых полиморфизмов выявил, что генотип AG полиморфизма A(-1903) гена химазы ассоциируется со стенокардией высокого ФК (р=0,104; рис. 2).
Установлено, что неблагоприятный генотип изучаемого полиморфизма гена химазы ассоциирован с наличием желудочковой экстрасистолии высоких градаций (р=0,013; рис. 3).
Обсуждение
 Разнообразие фенотипических проявлений у больных ГКМП обусловлено не только мутациями генов, кодирующих белки саркомера, но и другими генетическими факторами. Многие авторы изучали взаимосвязь полиморфизмов генов-модификаторов, характеризующих РААС, с некоторыми из клинических признаков ГКМП [8, 9].
Разнообразие фенотипических проявлений у больных ГКМП обусловлено не только мутациями генов, кодирующих белки саркомера, но и другими генетическими факторами. Многие авторы изучали взаимосвязь полиморфизмов генов-модификаторов, характеризующих РААС, с некоторыми из клинических признаков ГКМП [8, 9].
По нашему мнению, большой интерес представляло изучение ассоциации маркеров и полиморфизмов генов-модификаторов (AGT, AGTR1, CMA1, CYP11B2), характеризующих РААС, c клинико-гемодинамическими проявлениями, а также с клиническими вариантами течения.
Нами выявлено, что у больных ГКМП уровни АТII во много раз снижены по сравнению с таковыми в группе контроля (здоровые добровольцы). Таким образом, вместо ожидаемой активации циркулирующей РААС мы наблюдали снижение концентраций АТII в плазме крови. Стоит отметить, что заболевания, при которых содержание АТII повышено в циркулирующем русле, редки. Поэтому весьма важно понимание механизмов образования и действия АТII в тканях. Предполагается, что циркулирующий в крови АТII, главным образом, вовлечен в поддержание физиологического кровяного давления, тогда как тканевой АТII участвует в процессах атерогенеза и ремоделирования миокарда (рис. 4). Из-за того что циркулирующий АТII имеет доступ к тканям, трудно оценить истинные концентрации и эффекты тканевого АТII. Однако использование тканеспецифичных генетических моделей компонентов РААС помогло получить доказательство того, что локальные компоненты РААС ответственны за развитие тканевой патологии в значительно большей степени, чем циркулирующие [10].
Большинство органных систем экспрессируют все компоненты РААС, регуляция которых осуществляется независимо от циркулирующей системы. В экспериментальных исследованиях продемонстрировано, что даже в отсутствие АПФ концентрация АТII в тканях легких, сердца и почек остается неизменной [11]. Эти наблюдения указывают на важную роль альтернативных путей поддержания уровня АТII в тканях. Одним из таких путей является образование АТII при участии химазы [11].
По нашим данным, изучаемый полиморфизм гена химазы оказывал значимые влияния на маркеры РААС, что свидетельствует об активации тканевого звена РААС.
В то же время уровень АТII в кровяном русле был снижен. Исходя из этого можно допустить, что в процессах ремоделирования миокарда у больных ГКМП основную роль играют тканевые компоненты РААС. Подтверждением нашего предположения служат данные экспериментального исследования на трансгенных мышах, демонстрирующие, что гиперэкспрессия гена химазы приводит к 3-кратному повышению уровня АТII в тканях сердца, при том что в крови его уровень остается неизменным [12].
Можно предположить, что наибольшее количество АТII синтезируется в ткани сердца. В миокарде именно эта тканевая РААС способна к регулированию уровней АТII в пределах миокарда независимо от системного образования АТII [13, 14].
Кроме того, процессы ремоделирования миокарда и фиброза могут быть связаны не только с непосредственным влиянием АТII, синтезируемого при участии химазы, но и с эффектами альдостерона (гипертрофией миокарда, апоптозом кардиомиоцитов, синтезом коллагена, нарушением фибринолиза, усилением профибротического эффекта АТII) [15].
Другим изучаемым маркером у больных ССЗ является АПФ. Показано повышение активности АПФ в крови при таких заболеваниях, как инфаркт миокарда, диабетическая нефропатия, атеросклеротическое поражение сонных артерий и артериальная гипертония, что является свидетельством активации циркулирующего звена РААС [16—19].
P.C. Buck при изучении активности АПФ у больных семейной и спорадической формами ГКМП отметил корреляцию уровня АПФ с ИММЛЖ (р=0,04) [20]. Однако в нашем исследовании не обнаружено значимой связи АПФ ни с одним из показателей гипертрофии миокарда. Кроме того, не выявлено связи с показателями диастолической дисфункции, наличием пароксизмальной наджелудочковой тахикардии и стенокардии. При варианте прогрессирующего течения уровень маркера АПФ был выше, чем у больных с более благоприятными клиническими вариантами. Однако эти различия не были статистически значимыми.
Анализ основных синдромов ГКМП позволил установить наличие ассоциаций клинических проявлений заболевания с неблагоприятными генотипами изучаемых полиморфизмов, а также с маркерами РААС. Одним из ведущих признаков ГКМП является гипертрофия миокарда ЛЖ, которая обнаруживается у 92% больных с клиническими проявлениями заболевания. [21]. По данным литературы, гипертрофия ЛЖ ассоциируется с генотипом СС AGТ M235T [22, 23].
В нашем исследовании также обнаружена значимая связь данного генотипа со степенью гипертрофии миокарда. Помимо этого отмечена тенденция к увеличению ТЗСЛЖ у носителей неблагоприятного генотипа полиморфизма гена химазы. Диастолическая дисфункция — один из основных синдромов ГКМП. Однако мы не выявили ассоциаций изучаемых полиморфизмов генов-модификаторов с показателями диастолической дисфункции, что согласуется с исследованием L.A. Blauwet [24].

Полиморфизмы генов-модификаторов РААС рассматриваются как предикторы развития инфаркта миокарда и нестабильной стенокардии [25].
Помимо этого в нашей работе у больных ГКМП, которые являются носителями неблагоприятного генотипа изучаемого полиморфизма СМА1 (генотип AG), выявлено наличие таких нарушений ритма, как желудочковая экстрасистолия высоких градаций.
Таким образом, можно заключить, что генотип AG полиморфизма гена химазы СМА1 A(-1903)G является наиболее важным неблагоприятным фактором, оказывающим влияние на течение ГКМП: формирование гипертрофии миокарда, развитие нарушений ритма, преходящей ишемии миокарда.


