
Фиброзом называют уплотнение соединительной ткани в различных органах, которое сопровождается появлением рубцовых изменений. Эти рубцовые изменения возникают, как правило, на месте хронического воспаления: атрофии или дистрофии. В контексте фиброза основными «действующими лицами» являются компоненты внеклеточного матрикса — гликопротеины, протеогликаны и гиалуроновая кислота.
В пораженных тканях наблюдается чрезмерное накопление фибробластов и белков внеклеточного матрикса, включая коллаген, который служит основной структурной единицей фиброза. Коллаген — превалирующий гликопротеин внеклеточного матрикса. Это вещество представляет собой фибриллярный белок, который составляет основу соединительной ткани организма и обеспечивает ее прочность и эластичность. Пучки коллагена имеют форму тройной спирали, которые сшиваются вместе и образуют очень прочные коллагеновые фибриллы. Их прочность сравнима с прочностью стали [1].
Однако фиброз — не просто преобладание синтеза коллагена над его распадом. Это еще и процесс «структурирования» коллагена. Только структурированный коллаген имеет значение в развитии фиброза. Существуют 2 основных типа фиброза: реактивный и репаративный. Чаще всего мы встречаемся с реактивным типом, который наиболее характерен для большинства больных артериальной гипертонией (АГ), страдающих абдоминальным ожирением, с длительно текущей и плохо леченной АГ.
В качестве отсроченных последствий инфаркта миокарда можно рассматривать формирование на месте некроза рубца. Этот процесс является ярким примером репаративного фиброза. Атрофия и дистрофия ткани лежат в его основе. В этом случае пласты здоровых кардиомиоцитов чередуются с участками соединительной ткани [2].
Прогрессирование фиброза зависит, с одной стороны, от времени, а с другой — от микровоспалительной реакции, которая возникает в результате встречи с различными факторами (облучение, травма, инфекционно-аллергические факторы и др.). Выраженность микровоспалительной реакции определяет степень фиброза [3].
Конечно, в качестве одной из основополагающих причин фиброза нельзя не упомянуть значительную роль генетических структур, ответственных за синтез регуляторных белков. Эти белки участвуют в процессе фиброгенеза и могут приводить к ускорению или замедлению соответствующего процесса.
Главным механизмом в развитии фиброза служит активация ренин-ангиотензин-альдостероновой системы (РААС). Эта система контролирует тонус сосудов и гомеостаз натрия и воды. Основной действующий компонент РААС — ангиотензин-II (АТII), который представляет собой октапептид (состоит из 8 аминокислот). АТII образуется путем отщепления от неактивного предшественника ангиотензина-I (АТI) двух последних (C-концевых) аминокислот под воздействием ангиотензинпревращающего фермента (АПФ). АТII обладает высокой функциональной активностью, в первую очередь, в отношении сердечно-сосудистой системы, почек и надпочечников. Этот белок играет основную роль в развитии фиброза. Согласно данным экспериментальных и клинических исследований, ATII рассматривается как медиатор воспаления, который непосредственно активирует ремоделирование сердца. Кроме того, ATII отрицательно влияет на функцию эндотелия и стимулирует секрецию цитокинов и хемокинов. Но одним из наиболее отрицательных эффектов ATII в контексте фиброза служит его влияние на синтез коллагена. При этом ATII наравне с альдостероном снижает деградацию коллагена, что приводит к его выраженной аккумуляции.
 Эти эффекты АТII опосредуются взаимодействием с рецепторами АТII 1-го типа (АТ-1). Комплекс лиганд-рецептор активирует НАД-Н-оксидазу, образующую супероксид, который взаимодействует с вазорелаксирующим фактором (NO) и инактивирует его. Кроме того, АТII воздействует на корковое вещество надпочечников, стимулируя выделение альдостерона. Действие альдостерона приводит к задержке хлоридов и воды, усиленному выделению ионов Н+ и аммония, увеличению объема циркулирующей крови, а также сдвигу кислотно-основного состояния в сторону алкалоза. Действуя на клетки сосудов и тканей, гормон способствует транспорту Na+ и воды во внутриклеточное пространство [4].
Эти эффекты АТII опосредуются взаимодействием с рецепторами АТII 1-го типа (АТ-1). Комплекс лиганд-рецептор активирует НАД-Н-оксидазу, образующую супероксид, который взаимодействует с вазорелаксирующим фактором (NO) и инактивирует его. Кроме того, АТII воздействует на корковое вещество надпочечников, стимулируя выделение альдостерона. Действие альдостерона приводит к задержке хлоридов и воды, усиленному выделению ионов Н+ и аммония, увеличению объема циркулирующей крови, а также сдвигу кислотно-основного состояния в сторону алкалоза. Действуя на клетки сосудов и тканей, гормон способствует транспорту Na+ и воды во внутриклеточное пространство [4].
Итак, конечным результатом действия РААС является увеличение объема циркулирующей крови и повышение системного артериального давления (АД). Общие представления о РААС остались неизменными, однако появилась масса новых сведений. Обнаружено, что РААС не является исключительно циркуляторной системой, и в тканях различных органах, в частности, сердце, печени, поджелудочной железе, существует ее автономный аналог. В этих органах самостоятельно синтезируются отдельные фрагменты ангиотензина, которые участвуют в регуляции роста, апоптоза клеток, процессах воспаления и фиброгенеза.
Механизмы развития фиброза разных органов во многом схожи, но, учитывая особую интимную общность патогенетических реакций, приводящих к развитию фиброза сердца и печени, следует связать воедино эти два разных процесса.
К фиброзу миокарда приводят как гемодинамические, так и негемодинамические факторы. Одним из ключевых механизмов фиброза в данной ситуации является активация РААС. В ответ на механическое растяжение (например, при повышении системного АД) повышается давление внутри желудочка. Длительное растяжение стимулирует активность трансформирующего β-фактора роста (ТФР-β, TGF-β). Этот белок приводит к пролиферации фибробластов и накоплению компонентов межклеточного матрикса: коллагена, фибронектина, протео-гликанов. Одновременно начинается активная выработка АТII, который, воздействуя на рецепторы АТ-1, приводит к дополнительной стимуляции активности ТФР-β.
В результате увеличивается образование белков межклеточного матрикса и повышается организация коллагена [5].
Исходя из изложенного, можно сделать вывод, что к стимуляторам фиброгенеза относится не только АТII, но и ТФР-β. Существуют тесные связи между этими двумя составляющими. Эти связи не до конца раскрыты, однако интригующие результаты исследования J. Schultz и соавт. [6] показали, что АТII в отсутствии ТФР-β не может привести к фиброзу. В исследование были включены мыши с генетически детерминированным недостатком гена TGF-β. В условиях сниженного синтеза этого белка, несмотря на наличие циркулирующего АТII, у мышей отмечалось уменьшение фиброза и гипертрофии миокарда (рис. 1, см. цветную вклейку).
Действие АТII на миокард опосредуется через рецепторы 2 типов: АТII 1-го типа и АТII 2-го типа. Стимуляция этих рецепторов приводит к двум противоположным реакциям. Активация рецепторов АТII 1-го типа ведет к гипертрофии миокарда, накоплению белков внеклеточного матрикса и фиброзу. Стимуляция рецепторов АТII 2-го типа, напротив, ингибирует аналогичные процессы. Помимо классического, в сердце существует и автономный внутриорганный путь синтеза АТII. В условиях ингибирования АПФ повышается активность химазы. Этот фермент расщепляет в АТI связь между 8-й аминокислотой фенозином и 9-й аминокислотой гистидином. Образовавшийся АТII не гидролизуется дальше, так как связь между 4-й и 5-й аминокислотами в молекуле АТII устойчива к действию химазы. В результате при высвобождении химазы поддерживается не только постоянный уровень АТII, но и происходит образование сосудосуживающего пептида эндотелина-1. В итоге ремоделирование ткани прогрессирует [7].
Активация РААС, происходящая в рамках сердечно-сосудистой системы, влечет за собой не только изменения сердца, но и влияет на другие органы. Широко распространенное в общей популяции сочетание сердечно-сосудистых заболеваний (ССЗ) и болезней печени обладает общими патогенетическими звеньями [8].
Нетрудно обнаружить несколько параллелей в развитии фиброза печени и сердца. В качестве центрального звена также выступает АТII, который активирует ТФР-β. Резонный вопрос: как в печени появляется АТII? Печень пронизана сосудами, и изменения сосудов приводят к запуску процессов фиброгенеза. АТII активирует звездчатые клетки печени, которые трансформируются в миофибробласты — ключевое звено в патогенезе фиброза печени и сердца. Эти клетки опосредуют первичную выработку коллагена и накопление компонентов внеклеточного матрикса. В дальнейшем миофибробласты пролиферируют и высвобождают провоспалительные цитокины. Под воздействием АТII решающую роль в процессах фиброгенеза играют рецепторы АТ 1-го типа, которые преобладают в ткани печени [9].
Наряду с АТII к прогрессированию фиброза приводят также изменения базальной мембраны. В норме она представляет собой слой с выраженным перисинусоидальным пространством (просвет между стенками синусоидных капилляров и гепатоцитами в печеночной дольке). При механическом повреждении базальная мембрана теряет фенестрации, и обмен с кровотоком нарушается. Такая ситуация характерна для неалкогольной жировой болезни печени (НАЖБП), когда происходит жировое перерождение органа и, как следствие, изменяется уровень цитокинов, гормонов и нейротрансмиттеров.
В результате дезадаптации развиваются нейрогуморальные и иммунные нарушения. НАЖБП постепенно переходит в неалкогольный стеатогепатит (НАСГ), который является хроническим системным воспалительным процессом. Это усугубляет различные патологические процессы, лежащие в основе ССЗ. К ним относятся дисфункция эндотелия, атерогенез, тромбообразование, развивается дальнейшее повреждение сосудов [10].
Еще одним связующим звеном служат общие клинические предикторы развития фиброза печени и сердца [11]. К ним относятся следующие:
- возраст >45 лет;
- индекс массы тела (ИМТ) >31,1 кг/м2 (мужчины) и >32,3 кг/м2 (женщины);
- сахарный диабет 2-го типа (СД-2);
- АГ;
- повышение уровня С-пептида;
- аспартатаминотрансфераза (АсАТ)/аланинаминотрансфераза >1,
- АлАТ >2 нормальных значений;
- гипертриглицеридемия >1,7 ммоль/л.
Фиброз сердца и печени также имеет общую патофизиологическую основу, поэтому в данном случае нельзя не остановиться на одном из биомаркеров фиброза и неблагоприятного ремоделирования сердца – галектине-3. Впервые в журнале «Clinical Gastroenterology & Hepatology» было описано исследование, когда измеряли уровень галектина-3, который при НАЖБП оказался повышенным. Тогда же было выявлено, что при выключении генов, ответственных за синтез галектина-3, фиброз печени у мышей подвергался регрессу [12].
Завершающим звеном этой цепочки служит фиброз сосудов, который увеличивается с возрастом и практически всегда отмечается у пожилого человека. Фиброз сосудов — одна из основных причин стойкого повышенного АД. Стабильное повышение АД ведет к активации фибробластов и снижению активности металлопротеиназ [13].
Течение АГ у пациентов с НАЖБП имеет ряд особенностей. Для более детального изучения этого вопроса на кафедре пропедевтики внутренних болезней Первого МГМУ им. И.М. Сеченова проведено исследование, в котором участвовали 60 пациентов с АГ в сочетании с НАСГ (1-я группа) и 44 больных АГ без указаний на НАЖБП (2-я группа). Результаты исследования показали, что для больных АГ в сочетании с болезнями печени более характерны увеличение печени, одышка, тахикардия. Значительно превышали норму уровни АлАТ и АсАТ. Кроме того, в ходе работы показано, что у пациентов с заболеваниями печени отмечается более тяжелая АГ с более высокими уровнями систолического (САД) и диастолического (ДАД) АД по сравнению с таковыми у больных без заболеваний печени. У больных АГ в сочетании с НАСГ также отмечались более высокие уровни показателей, отражающих инсулинорезистентность (ИР): уровень глюкозы в крови, инсулина и С-пептида. Кроме того, в группе больных со стеатогепатитом уровни инсулина, глюкозы и С-пептида тесно коррелировали с ИМТ. Выявлены также положительные корреляции между уровнями трансаминаз и показателями ИР, а именно АсАТ и С-пептидом, АсАТ и инсулином, АлАТ и С-пептидом. Полученные данные свидетельствуют о том, что ИР играет центральную роль в развитии заболеваний печени и прогрессировании ССЗ у пациентов с АГ в сочетании с НАСГ [14].
 Отрицательное влияние ИР на печень заключается в избыточном образовании свободных жирных кислот, что приводит к отложению жира в печени. Накопление жира в гепатоцитах ведет к развитию стеатоза и липотоксичности. Помимо этого свободные жирные кислоты подвергаются окислению, что приводит к ингибированию К/Na-АТФазы, угнетению гликолиза, разобщению окислительного фосфорилирования. Защитные свойства мембраны гепатоцитов резко снижаются, что приводит к повреждению митохондрий, апоптозу и некрозу гепатоцитов.
Отрицательное влияние ИР на печень заключается в избыточном образовании свободных жирных кислот, что приводит к отложению жира в печени. Накопление жира в гепатоцитах ведет к развитию стеатоза и липотоксичности. Помимо этого свободные жирные кислоты подвергаются окислению, что приводит к ингибированию К/Na-АТФазы, угнетению гликолиза, разобщению окислительного фосфорилирования. Защитные свойства мембраны гепатоцитов резко снижаются, что приводит к повреждению митохондрий, апоптозу и некрозу гепатоцитов.
Учитывая, что фиброз развивается и в сердце, и в печени, важно подобрать препарат, который вмешивался бы в процессы фиброза в обоих органах, но при этом обладал минимальной гепатотоксичностью, не требовал дополнительной метаболизации в печени и циркулировал в плазменном звене РААС [15].
Наиболее оправданным у таких пациентов является комплексный подход к лечению, включающий применение антигипертензивной терапии, препаратов, направленных на улучшение функции печени (эссенциальные фосфолипиды, урсодезоксихолевая кислота, адеметионин и др.), гиполипидемических средств (статины), а при необходимости и гипогликемических препаратов (метформин). Такое лечение оказывает достоверный положительный эффект и приводит к нормализации суточного АД, липидного обмена и гликемии [16].
Показано, что широко используемые в настоящее время препараты, воздействующие на РААС посредством ингибирования АПФ или блокирования ангиотензиновых рецепторов, могут вмешиваться в процессы фиброза в обоих органах.
У больных АГ в сочетании с НАСГ на первый план выходит хорошо изученный класс препаратов ингибиторов АПФ, которые давно зарекомендовали себя в качестве безопасных и эффективных средств [17].
Существует большое количество ингибиторов АПФ, все они способны вмешиваться в процессы фиброза как в сердце, так и в печени. Эти препараты различаются по гидро- и липофильности, продолжительности действия, фармакокинетике. Однако лечение АГ у пациентов высокого риска сердечно-сосудистых осложнений требует проведения комбинированной антигипенртензивной терапии. Именно поэтому у пациентов с АГ и НАСГ определенную пользу можно ожидать от комбинированного антигипертензивного препарата, содержащего лизиноприл и амлодипин (экватор, Гедеон Рихтер, Венгрия). Комбинированная терапия амлодипином и лизиноприлом оказывает выраженный гипотензивный эффект и отличается хорошей переносимостью у больных с умеренной и тяжелой АГ, а также с нарушением функции печени. Более того, данная комбинация улучшает диастолическую дисфункцию левого желудочка (ЛЖ) [18, 19]. Сочетание амлодипина и лизиноприла заметно превосходит по антигипертензивному эффекту каждый компонент, применяемый в отдельности.
Во многих клинических ситуациях данная комбинация служит надежным помощником в комплексной терапии у пациента с избыточной массой тела, страдающего АГ и НАЖБП. Нетрудно догадаться, что это портрет пациента с метаболическим синдромом (МС). Для оценки эффективности и безопасности применения экватора у пациентов с МС было спланировано и проведено клиническое исследование ДИРИЖЕР («ДИРотон в лечении артерИальной гипертензии у пациентов с метаболическим синдромом и неалкогольной Жировой болЕзнью печени»).
В исследование включали пациентов в возрасте от 18 до 65 лет с МС, АГ и сопутствующей НАЖБП. Выбраны следующие критерии включения:
- мужчины и женщины;
- возраст старше 18 и моложе 65 лет;
- САД 140 мм рт.ст. и более и/или ДАД 90 мм рт.ст. и более;
- наличие МС: абдоминальное ожирение (окружность талии — ОТ >94 см у мужчин, >80 см — у женщин) и любые 2 других критерия МС: уровень глюкозы в крови натощак более 100 мг/дл, низкий уровень холестерина (ХС) липо-протеидов высокой плотности — ЛВП (≤40 мг/дл у мужчин, ≤50 мг/дл у женщин), высокий уровень триглицеридов — ТГ (>150 мг/дл);
- сопутствующее заболевание печени в виде НАЖБП (данные ультразвукового исследования (УЗИ), АсАТ/АлАТ, маркеры вирусных инфекций);
- отмена ранее проводимой антигипертензивной терапии за 7 сут до включения больных в исследование.
Из 100 пациентов с АГ критериям включения соответствовали 72. Средний возраст пациентов, включенных в исследование, составлял 50,6±12,6 года, среди пациентов было 45% женщин (n=32) и 55% мужчин (n=40).
Все пациенты с МС в качестве обязательной составляющей имели абдоминальное ожирение со средним ИМТ 33,1±5,3 кг/м2 и ОТ 110,7±11,4 см.
У 100% пациентов диагностирована НАЖБП, из них у 64% — стеатоз печени, у 36% (n=26) — НАСГ. Отмечалось умеренное повышение уровня АсАТ до 45±37 ед/л и АлАТ до 40±32 ед/л.
В качестве сопутствующих заболеваний у 11,1% пациентов имелся СД-2, у 83,3% — атерогенная дислипидемия.
Пациенты были рандомизированы в 2 группы: в 1-й группе (n=38) пациенты применяли комбинацию препаратов на основе лизиноприла, во 2-й (n=34) — на основе периндоприла.
Признаки ИР отмечались у всех больных, включенных в исследование.
Пациенты по показаниям получали сопутствующую терапию статинами.
Период наблюдения составлял 12 нед. Проведено 4 визита. Пациенты обследованы исходно и через 12 нед терапии. Методы исследования включали клинический анализ крови; биохимическое исследование плазмы (липидный состав крови: общий холестерин – ОХС, ХС липопротеидов низкой плотности — ЛНП, ХС ЛВП, ТГ; АлАТ, АсАТ, К+, глюкоза, креатинин); общий анализ мочи; инсулин крови, тест на ИР НОМА; электрокардиограмма в 12 отведениях; УЗИ органов брюшной полости; ультразвуковая допплерография сонных артерий Проводились: суточное мониторирование АД; эхокардиография — ЭхоКГ (оценка гипертрофии миокарда, сократительной функции ЛЖ, оценка диастолической функции).
Исходные параметры ЭхоКГ позволили выявить наличие у пациентов сохранной фракции выброса ЛЖ 62±4%. Масса миокарда ЛЖ составила 219±58 г, индекс массы миокарда ЛЖ 106,9±19,3 г/м2, толщина межжелудочковой перегородки 1,15±0,16 см, задняя стенка ЛЖ 1,1±0,12 см, что указывало на гипертрофию миокарда ЛЖ, незначительную и умеренную дилатацию левого предсердия (ЛП), объем ЛП составил 59±15 мл. Средняя толщина эпикардиальной жировой ткани составляла 0,5±0,1 см.
Средние показатели диастолической функции ЛЖ у пациентов до лечения указывали на наличие диастолической дисфункции по I типу — нарушение релаксации ЛЖ. Регистрировались уменьшение пика Е и увеличение пика А по трансмитральному потоку, уменьшение е′ и увеличение а′ по результатам тканевой допплерографии фиброзных колец митрального клапана. Умеренно выраженная диастолическая дисфункция также подтверждалась соотношением Е/А = 0,69±0,08 и соотношением E/е′ = 7,9±0,9 (показатель E/е′ >7,0 выявлялся у всех пациентов с МС).
При УЗИ внечерепных отделов сонных артерий обращало внимание увеличение толщины интимы—медии (ТИМ) до 1,08±0,068 мм. Атеросклеротические бляшки в сонных артериях выявлялись у 20 (27,8%) больных.
При статистическом анализе использовали программу Statistica 7.0. Вычислены описательные статистики, критерий χ2 и корреляции между переменными, построены таблицы сопряженности. Сравнение выборок реализовано методами непараметрической статистики, дисперсионного анализа, для сравнения независимых переменных использовали критерии Манна—Уитни и Стьюдента с оценкой их значимости р. За уровень достоверности статистических показателей принято р<0,05.
Результаты и обсуждение
По данным суточного мониторирования АД, у пациентов с избыточной массой тела, НАЖБП и МС выявлены особенности клинического течения АГ. Констатировано наличие систолодиастолической АГ в дневные часы с более выраженным повышением САД, систолической АГ в ночные часы, повышение индексов гипертонической нагрузки в дневные и ночные часы по САД и ДАД, дополнительное повышение пульсового АД, нарушение суточного колебания АД с недостаточным снижением САД в ночные часы. Такие данные свидетельствуют о более высоком риске поражения органов-мишеней у больных с МС и требуют назначения высокоэффективных препаратов.
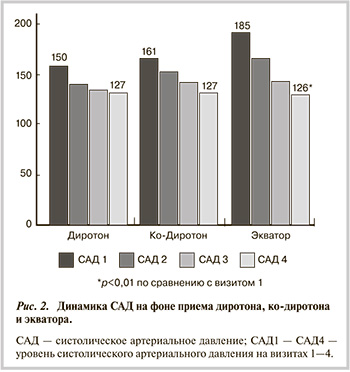
В группе антигипертензивной терапии на основе лизиноприла достигнуты целевые уровни АД во всех подгруппах (рис. 2, 3). Причем на фоне фиксированной комбинации лизиноприла с амлодипином отмечалось максимальное снижение САД и ДАД.
На фоне терапии через 12 нед отмечался регресс гипертрофии миокарда ЛЖ с 225 до 214 г, уменьшился объем ЛП c 61 до 55 мл в группе лизиноприла и с 56 до 51 мл в группе периндоприла (р<0,01).
На фоне обоих режимов антигипертензивной терапии отмечалось улучшение диастолической функции ЛЖ. Увеличилось отношение Е/А в обеих группах, а также уменьшилось отношение E/е′.
Регресс гипертрофии миокарда ЛЖ, уменьшение размеров ЛП, улучшение диастолы предполагают профилактику ремоделирования сердца. Известно, что ремоделирование — это структурно-геометрическое изменение ЛЖ, включающее процессы гипертрофии миокарда и дилатации полостей сердца, что ведет к изменению его геометрии, нарушению систолической и диастолической функции.
Следует отметить, что несмотря на эффективную антигипертензивную терапию и сопутствующую терапию статинами, ТИМ достоверно не изменилась через 12 нед терапии, средняя ТИМ до лечения составляла 1,08±0,068 мм, через 3 мес — 1,078±0,06 мм (р>0,05). Для регресса процессов атеросклероза требуется более длительное наблюдение. Кроме того, на фоне сопутствующей терапии статинами отмечалось улучшение показателей липидного состава крови и ИР у всех пациентов.
Таким образом, результаты проведенного исследования продемонстрировали, что применение комбинированной терапии ингибитором АПФ и амлодипина (экватор) у пациентов с МС приводит к снижению САД и ДАД; уменьшению гипертрофии ЛЖ, размеров предсердий; к улучшению диастолического расслабления.
Следует отметить хорошую переносимость данной терапии. Побочные эффекты в виде отеков ног не были причиной отмены препаратов в данном исследовании [19].
Заключение
Не остается никаких сомнений в том, что сердечно-сосудистые заболевания и болезни печени связаны между собой и, более того, усугубляют течение друг друга. Развитие фиброза в печени, сердце и сосудах имеют общий механизм — активацию ренин-ангиотензин-альдостероновой системы. Это усиливает исследовательский интерес к гепатокардиальным связям. Данная связь прослеживается и у больного артериальной гипертонией с избыточной массой тела, и у пациентов с алкогольной зависимостью, и у больных с сердечной недостаточностью. Детализация механизмов развития гепатокардиальной оси крайне увлекательна и многогранна. Поиск препаратов, влияющих на развитие фиброза, переводит эту область из сферы исследовательского интереса в плоскость практической медицины.


