ВВЕДЕНИЕ
Гепатоцеллюлярный (печеночноклеточный) рак (ГЦР) – наиболее частая (порядка 85% случаев) первичная злокачественная опухоль печени, происходящая из гепатоцитов. Ежегодно ГЦР выявляется у более чем 900 тыс. человек во всем мире [1]: это 6-й по частоте встречаемости вид рака и 4-я по значимости причина онкологической смертности [2]. В России ГЦР встречается сравнительно редко, однако стандартизованная заболеваемость с 2010 по 2022 г. выросла на 36 и 57% среди мужчин и женщин соответственно [3], что отражает общемировую тенденцию [1].
ГЦР чаще всего (около 80% случаев) развивается на фоне цирроза печени или хронического воспаления любой этиологии: вирусных гепатитов В (ВГB) и С (ВГC), алкогольного и неалкогольного стеатогепатита (НАСГ), вследствие экзогенных токсических повреждений печени, а также при некоторых наследственных заболеваниях и нарушениях состояния иммунной системы [4]. Хотя стратификация случаев ГЦР по этиологии значительно варьируется между странами и континентами, преобладающими причинами в большинстве регионов являются ВГB и ВГC [2]. Тем не менее на фоне роста охвата вакцинацией против ВГB и доступности эффективных препаратов для лечения ВГC возрастает значимость НАСГ как этиологического фактора ГЦР. Так, в США ВГC по-прежнему остается ведущей этиологией ГЦР у мужчин, однако у женщин с 2017 г. 1-е место среди причин стабильно занимает НАСГ [5]. Ожидается, что именно НАСГ станет преобладающей причиной ГЦР в странах с высокими уровнями развития и доходов населения [2].
ГЦР характеризуется агрессивным течением и неблагоприятным прогнозом: средняя 5-летняя выживаемость при этой патологии не превышает 20% [6], а 5-летняя частота рецидивов даже после радикального хирургического лечения достигает 70% [7]. Ввиду сложности своевременной диагностики и успешного лечения ГЦР актуальна проблема его первичной и вторичной профилактики в популяциях пациентов высокого риска. Результаты трансляционных и клинических исследований позволили сформулировать различные стратегии общей и этиологически специфичной хемопревенции ГЦР, направленные на коррекцию хронического воспаления, метаболической дисфункции, оксидативного стресса и фиброза печени как доказанных факторов канцерогенеза [7]. Несмотря на многочисленность лекарственных средств и биодобавок, рассматриваемых в качестве хемопревентивных агентов, возможности их применения в рутинной клинической практике ограничены малым объемом и несистематическим характером доказательств их эффективности и безопасности [8].
Адеметионин (S-аденозил-L-метионин, SAM) – эндогенная аминокислота, обладающая гепатопротекторным, детоксицирующим, антихолестатическим, умеренным холеретическим и антидепрессивным действием. Известные механизмы действия адеметионина включают прямое участие в биосинтетических реакциях, работе антиоксидантных систем и детоксификации желчных кислот и ксенобиотиков. Помимо этого, в последнее время все большее внимание уделяется роли адеметионина как прямого регулятора процессов апоптоза и аутофагии, что опосредует его собственный противоопухолевый эффект и синергизм со многими антинеопластическими агентами [9]. Оригинальный адеметионин в исследованиях демонстрировал эффективность при лечении гепатотоксичности, обусловленной химиотерапией [10].
В настоящем обзоре рассмотрены механизмы фармакологических эффектов адеметионина в контексте его потенциальной хемопревентивной и противоопухолевой активности при ГЦР.
БИОСИНТЕЗ SAM
В печени SAM синтезируется путем реакции между АТФ и метионином, катализируемой преимущественно изоформами I и III метионинаденозилтрансферазы (MAT). MAT-I/III экспрессируется в печени взрослого организма, считается маркером дифференцированного клеточного фенотипа и антионкогеном [11].
При тяжелых повреждениях и циррозе органа наблюдается снижение активности печеночной MAT-I/III на 50–60%, что объясняется как угнетением ее экспрессии (в том числе вследствие метилирования ДНК), так и посттрансляционной ковалентной инактивацией оксидом азота и гидроксильными радикалами. Менее выраженное, но значительное снижение активности MAT-I/III и, как следствие, уменьшение уровня SAM могут присутствовать и у пациентов с прецирротическими стадиями алкогольной болезни печени (АБП) [12]. Нокаут гена MAT-I/III и дефицит SAM предрасполагали мышей к развитию оксидативного стресса и стеатоза печени со спонтанным прогрессированием до стеатогепатита и ГЦР [13]. Напротив, трансфекция MAT-I в клетки ГЦР Huh7 замедляла рост и васкуляризацию опухолей, образованных ими после введения мышам [14].
Одновременно при поражении печени происходит транскрипционная индукция MAT-II, которая в норме является внепеченочной изоформой и реализует отличные от MAT-I/III свойства. MAT-II имеет значительно меньшее сродство к субстрату, принимает незначительное участие в синтезе SAM, но стимулирует синтез ДНК, быструю пролиферацию и дедифференциацию клеток печени. Поскольку SAM служит естественным ингибитором MAT-II, уменьшение его продукции способствует поддержанию активности этой изоформы [13]. Повышение печеночной экспрессии MAT-II отмечено в гепатоцитах, пораженных вирусом гепатита С [15], при частичной гепатэктомии [16], поражении печени тиоацетамидом [17] или этанолом [18] у крыс, а также при ГЦР у человека [19].
УЧАСТИЕ В БИОСИНТЕТИЧЕСКИХ ПРОЦЕССАХ
SAM в качестве кофактора вовлечен в множество анаболических реакций, традиционно объединяемых в пути трансметилирования, транссульфирования и аминопропилирования [11]. К основным первичным реакциям метаболизма SAM относятся деметилирование под действием метилтрансфераз и декарбоксилирование посредством декарбоксилазы SAM.
Метильная группа в составе SAM является химически активной и может переноситься посредством метилтрансфераз на широкий спектр субстратов, включая фосфолипиды, углеводы, гормоны, нейромедиаторы-моноамины, нуклеиновые кислоты и гистоны. Стимуляция синтеза фосфатидилхолина и стабилизация мембран гепатоцитов лежит в основе антицитолитического эффекта SAM, наблюдавшегося в исследованиях при НАСГ [20] и поражениях печени противотуберкулезными препаратами [21].
Декарбоксилированный SAM расходуется в реакциях аминопропилирования с образованием полиаминов спермина и спермидина, принимающих участие в регуляции транскрипции, трансляции и клеточного цикла [11]. Оба этих соединения необходимы для пролиферации гепатоцитов и регенерации ткани печени после ее повреждения [22]. При синтезе спермина и спермидина как побочный продукт образуется метилтиоаденозин (MTA), который может превращаться в метионин и впоследствии в исходный SAM [11].
Спермин способен изменять характер поляризации клеток Купфера и уменьшать продукцию ими провоспалительных цитокинов, что опосредует его противовоспалительное и антифибротическое действие при острых токсических поражениях печени [23]. Тем не менее известно, что спермин играет важную роль в поддержании биосинтетических процессов, антиоксидантной защите и межклеточных взаимодействий клеток ГЦР, а также опосредует уклонение опухоли от иммунного надзора [24]. Кроме этого, для больных ГЦР характерна гиперактивация сперминсинтазы, которая катализирует SAM-зависимое превращение спермидина в спермин [25].
Спермидин и его производные, напротив, стимулируют иммунный ответ на опухолевые антигены, повышают стабильность микротрубочек и предупреждают онкогенез путем индукции аутофагии или апоптоза клеток [26]. Экзогенный спермидин оказывал положительное влияние на течение ишемически-реперфузионного [27] и алкогольного [28] поражения печени, однако не улучшал морфологическую картину печени при экспериментальном НАСГ [29]. У мышей с тетрахлорметан-индуцированным поражением печени спермидин замедлял фиброгенез, снижал риск развития ГЦР и увеличивал общую продолжительность жизни [30]. В условиях in vitro он обладал прямой активностью в отношении клеток ГЦР, а также продемонстрировал выраженный синергизм с иммунодепрессантом ауранофином [31]. В связи со сказанным сложно однозначно определить, какой вклад участие SAM в биосинтезе полиаминов вносит в его потенциальную противоопухолевую активность.
АНТИОКСИДАНТНОЕ ДЕЙСТВИЕ
После деметилирования SAM превращается в S-аденозилгомоцистеин (SAH), который включается в серию последовательных реакций транссульфирования, приводящих к образованию тиолсодержащей аминокислоты цистеина. Цистеин выступает предшественником восстановленного глутатиона, сероводорода и таурина – важнейших эффекторов клеточной антиоксидантной системы. Кроме этого, цистеин необходим для образования кофермента А и железосерных кластеров, выполняющих кофакторную функцию во многих окислительно-восстановительных реакциях [11]. SAM обладает невысокой собственной скэвенджерной активностью, но может принимать участие в хелатировании ионов Fe2+ и ингибировать их аутоокисление, что усиливает его антиоксидантный потенциал [32].
Оксидативный и нитрозативный стресс, возникающий на фоне хронического воспаления печени, является фактором образования ДНК-аддуктов, дисрегуляции экспрессии про- и антионкогенов, укорочения и дестабилизации теломер, нарушения репарации ДНК и ингибирования апоптоза клеток. Использование прямых или косвенных антиоксидантов (токоферола, куркумина, глицирризиновой и урсодезоксихолевой кислот и др.) для уменьшения выраженности оксидативного стресса и его последствий рассматривается как одно из перспективных направлений хемопревенции у больных НАСГ, вирусными гепатитами и циррозом печени [33]. Менее однозначной представляется целесообразность применения антиоксидантов при уже развившейся ГЦР, поскольку антиоксиданты могут облегчать ее рост и метастазирование, а также снижать эффективность мультикиназных ингибиторов (сорафениба и др.) [34, 35], используемых в качестве средств 1–2-й линий у больных ГЦР [4].
В экспериментах in vitro SAM снижал выраженность оксидативного стресса в клетках ГЦР FaO и эндотелиоцитах HECV, подвергнутых перегрузке высшими жирными кислотами, уменьшая проявления стеатоза и эндотелиальной дисфункции [36]. В клетках Huh7, несущих субгеномный РНК-репликон ВГС, SAM индуцировал супероксиддисмутазы-1/2 и тиоредоксин-1, что сопровождалось снижением уровня вирусной РНК на 50–70% [15]. У мышей введение SAM через час после интоксикации парацетамолом уменьшало проявления оксидативного стресса и образование маркерных парацетамол-белковых аддуктов [37]. Кроме этого, на мышах, трансгенных по белку-прекурсору амилоида, было показано, что SAM и супероксиддисмутаза обладают выраженным синергизмом при пероральном введении [38].
АНТИХОЛЕСТАТИЧЕСКОЕ ДЕЙСТВИЕ
Гидрофобные желчные кислоты (дезоксихолевая, хенодезоксихолевая, таурохенодезоксихолевая, литохолевая) обладают прооксидантными свойствами, повреждают ДНК и снижают геномную стабильность, способствуя опухолевому перерождению гепато- и холангиоцитов. Избыток желчных кислот изменяет микроокружение опухоли, облегчая ее уклонение от иммунного ответа, инвазию и метастазирование [39].
Антихолестатический эффект SAM связывают со стимуляцией биосинтеза мембранных фосфолипидов, а также с образованием в ходе транссульфирования таурина, участвующего в конъюгации желчных кислот с образованием малотоксичных и легко элиминируемых из организма таурохолатов [40]. Помимо этого, таурин обладает мембранопротекторной и осморегулирующей активностью, связывает токсичные катионы тяжелых металлов и гипохлорную кислоту. На животных моделях АБП, неалкогольной жировой болезни (НАЖБП) и фиброза печени экзогенный таурин уменьшал проявления оксидативного стресса, воспаление и стеатоз гепатоцитов, способствовал конверсии холестерина в желчные кислоты и детоксификации последних [40].
Таурин ингибировал пролиферацию и индуцировал апоптоз клеток HepG2 [41], повышал противоопухолевую активность сорафениба и снижал его иммунотоксичность in vitro [42], а также показал потенциальный иммуностимулирующий эффект при применении в комбинации с куркумином и пиперином у больных нерезектабельным местнораспространенным или метастатическим ГЦР [43].
АНТИФИБРОТИЧЕСКОЕ ДЕЙСТВИЕ
Тяжелый фиброз и цирроз печени – ведущий фактор риска развития ГЦР, в связи с чем использование антифибротических средств рассматривается как основная стратегия ее хемопревенции [7]. Безусловную важность в контексте антифибротической активности SAM имеют его описанные антиоксидантные свойства. В то же время данные экспериментальных исследований позволяют предположить наличие и иных механизмов ингибирования фиброгенеза SAM.
Субъединицы α2 и β MAT-II, кодируемые генами Mat2a и Mat2b соответственно, играют важную роль в активации звездчатых клеток печени (ЗКП) и запуске процессов фиброгенеза под влиянием трансформирующего фактора роста-β1 (TGF-β1) [44] и лептина [45]. TGF-β1 в ЗКП и опухолевых клетках активирует транскрипцию субъединицы p65 ядерного фактора-κB (NF-κB), которая, в свою очередь, является прямым индуктором Mat2A [44]. Отметим, что активация NF-κB характерна практически для всех видов хронических поражений печени, включая АБП, НАЖБП, холестатические патологии и вирусные гепатиты [46].
Снижение клеточных уровней SAM вследствие нарушения равновесия MAT-I/II приводит к гипометилированию ДНК, служащему ключевым механизмом эпигенетического регулирования экспрессии генов и трансдифференциации ЗКП [47]. Сайленсинг Mat2A, напротив, сопровождается замедлением активации и роста первичных ЗКП [48]. Экзогенный SAM снижал базальную и TGF-β1-индуцированную транскрипцию MAT-II, проколлагена-А1 и α-актина гладких мышц в активированных ЗКП [44]. Введение SAM мышам сопровождалось индукцией фактора Smad7, который выполняет роль не только транскрипционного ингибитора TGF-β1, но и косвенного блокатора NF-κB [49, 50]. Индуктор Smad7 пирфенидон демонстрирует высокую активность в отношении идиопатического легочного [51], тубулоинтерстициального [52] и печеночного [53] фиброза, а его производное гидронидон (в комбинации с энтекавиром) успешно прошло клиническое испытание II фазы с участием больных фиброзом печени, вызванным ВГB [54].
Клеточные эффекты лептина зависят от экспрессии как Mat2a, так и Mat2b. При этом нокдаун Mat2b в клетках HepG2 препятствовал активации мишеней лептина STAT3 (сигнальный белок и активатор транскрипции-3), ERK (киназа, регулируемая внеклеточным сигналом) и PI3K (фосфатидилинозитол-3-киназа), опосредующих его профиброгенный эффект. SAM уменьшал митогенное действие лептина в культуре HepG2, не влияя на активность STAT3/ERK/ PI3K, в то время как его метаболит MTA ингибировал их [45]. Неизвестно, могут ли SAM и/или MTA влиять на эти сигнальные молекулы в ЗКП, или их антифибротический эффект, показанный на моделях in vivo [55], реализуется посредством иных механизмов. Значение для антифибротического потенциала SAM может иметь его влияние на сигнальные пути Wnt/β-катенина, индуцируемые в том числе лептином [56].
В экспериментах на изолированных ЗКП SAM подавлял синтез коллагена I [57], стимулировал его распад путем убиквитинилирования [58], а также препятствовал сокращению клеток, нарушая образование филаментов F-актина и миозина [59]. У крыс SAM (3 мг на килограмм в день) ингибировал печеночную пролилгидроксилазу и уменьшал продукцию коллагена, вызванную воздействием тетрахлорметана, а также снижал риск прогрессии состояния до цирроза печени [55]. MTA в большей степени, нежели SAM, снижал экспрессию генов TGF-β1, TGF-α и проколлагена-А1 в печени крыс, подвергнутых хронической интоксикации тетрахлорметаном [60].
ЭПИГЕНЕТИЧЕСКИЕ ЭФФЕКТЫ
SAM необходим для функционирования всех известных ДНК-метилтрансфераз, переносящих метильную группу на остатки цитозина. При этом SAH, образующийся в результате этого переноса, конкурентно ингибирует ДНК-метилтрансферазы [11]. Отношение уровней SAM и SAH получило название индекса метилирования, отражающего интенсивность клеточного метилирования ДНК – одного из основных эпигенетических механизмов регуляции экспрессии генов. Нарушения профиля метилирования ДНК и последующие изменения протеома клетки считаются важным фактором патогенеза хронических заболеваний печени, включая алкогольную и неалкогольную жировую болезни, а также их прогрессирования до фиброза и цирроза печени [61, 62]. Поскольку аномальные паттерны метилирования и деметилирования ДНК также являются ранним событием в канцерогенезе, они рассматриваются как потенциальные биомаркеры для выявления ГЦР на начальных стадиях [63].
В экспериментах на животных было установлено, что дефицит SAM, вызванный этионином, этанолом, метионин- и/или холиндефицитными диетами, приводит к деметилированию и активации промоторов онкогенов, вовлеченных в процессы пролиферации, клеточной адгезии и метастазирования [64]. В последующих работах была подтверждена гипотеза, что восполнение этого дефицита способствует восстановлению нормального профиля метилирования ДНК, тем самым предупреждая или замедляя канцерогенез. Экзогенный SAM оказывал дифференциальное влияние на транскриптом опухолевых клеток печени с различным фенотипом (неинвазивных HepG2 и высокоинвазивных SKhep1), в то же время значительно слабее воздействуя на нормальные гепатоциты. При этом увеличение уровня SAM, как возможно было ожидать, не сопровождалось неизбирательным общим повышением степени метилирования ДНК, а приводило к селективному гипер- или гипометилированию различных генов, что приближало параметры метилома опухолевых клеток к нормальным [64].
В клетках ГЦР HepG2 и HuH7 SAM угнетал экспрессию циклина-D1, регулирующего фазовый переход G1/S, фактора пролиферации и лекарственной устойчивости клеток ГЦР E2F1, провоспалительных факторов IKK (киназа IκB) и NF-κB (ядерный фактор-κB), ингибиторов апоптоза BCL2 и XIAP, а также, напротив, индуцировал проапоптотические белки Bax и Bak [65]. SAM эффективно подавлял экспрессию урокиназного активатора плазминогена (uPA) и матричных металлопротеиназ (MMP) в клетках остеосаркомы (LM-7, MG-63) и высокоинвазивных опухолей молочной железы (MDA-231), простаты (PC-3) и толстой кишки (SW-620) [66]. Гиперэкспрессия uPA и активируемых им MMP опосредует эпителиально-мезенхимальный переход и стимулирует неоангиогенез, инвазивный рост и метастазирование ГЦР [67, 68].
В клетках колоректальной аденокарциномы SAM индуцировал экспрессию гистона H2AX, ассоциированного с активацией процессов репарации ДНК, а также уменьшал признаки нестабильности генома [69]. Ингибированию промоторов фактора роста эндотелия сосудов-C (VEGF-C) под влиянием SAM сопутствовало значительное замедление роста различных линий клеток рака желудка [70]. Кроме этого, в клетках карцином желудка и толстой кишки SAM восстанавливал метилирование промоторов онкогенов c-myc и H-ras, в то же время не влияя на экспрессию онкосупрессора p16 [71].
Обнаружено, что восстановление профиля метилирования ДНК позволяет SAM действовать синергично с деметилирующим агентом децитабином [72], антиметаболитом 5-фторурацилом [73] и ингибиторами контрольных точек иммунного ответа (пембролизумаб, ниволумаб и др.) [74], обладающими клинической эффективностью при ГЦР [75, 76].
ДРУГИЕ ПРОТИВООПУХОЛЕВЫЕ ЭФФЕКТЫ
Вероятным промежуточным механизмом антионкогенного действия SAM может выступать ингибирование митогенного эффекта фактора роста гепатоцитов (HGF) [77] с преимущественным воздействием на его AMPK-ветвь [78]. Тем не менее некоторые экспериментальные данные указывают на то, что SAM способен ингибировать и ветвь ERK, а также сигналинг протеинкиназы B (Akt). Именно с этим механизмом, вероятно, был связан его синергизм с селеносодержащими соединениями (селенометионином, метилселеноцистеином, метилселеновой кислотой), обладающими собственной противоопухолевой активностью [79]. Ингибиторы Akt (мирансертиб, веворисертиб) являются эффективными модуляторами опухолевого микроокружения, благодаря чему рассматриваются как потенциальные средства терапии солидных опухолей, включая ГЦР [80].
SAM и его продукт MTA способны повышать экспрессию протеинфосфатазы-1 (PP1), которая модулирует альтернативный сплайсинг регулятора апоптоза Bcl-x. В клетках ГЦР SAM и MTA сдвигают соотношение сплайс-вариантов мРНК гена Bcl-x в сторону преобладания Bcl-xS (малой изоформы), обладающей проапоптотическими свойствами [81]. Кроме этого, SAM и MTA подавляют транскрипцию бетаин-гомоцистеин-S-метилтрансферазы (BHMT), тем самым индуцируя стресс эндоплазматического ретикулума и апоптоз опухолевых клеток [82]. Следует отметить, что оба эффекта не наблюдаются в нормальных гепатоцитах [81, 82].
МТА как побочный продукт биосинтеза полиаминов ингибирует скорость-лимитирующие ферменты орнитиндекарбоксилазу (ODC) и декарбоксилазу SAM, гиперэкспрессия которых характерна для клеток печени на пренеопластической и неопластической стадиях [83]. Поскольку индукция ODC происходит раньше, чем значимые нарушения синтеза ДНК в перерождающихся клетках, ингибиторы ODC (эфлорнитин) применяются для хемопревенции у онкобольных высокого риска. Обсуждается возможность применения ингибиторов ODC и малых интерферирующих РНК в качестве средств основной или адъювантной терапии ГЦР [84].
В различных видах опухолей SAM, по-видимому, может реализовывать свой эффект различными путями. Так, в клетках колоректальной карциномы он угнетал продукцию циклина-D1, ускорял расщепление поли(АДФ-рибоза)-полимеразы-1 (PARP1), блокировал клеточный FLICE-подобный ингибиторный белок (cFLIP) и повышал активность проапоптотических каспаз [66, 85]. Активация каспаз-3 и -8 обусловливала синергизм SAM с доксорубицином в отношении клеток гормонозависимого рака молочной железы CG5 и MCF-7 [86], однако может быть благоприятным эффектом и в условиях ГЦР [87].
Гиперэкспрессия циклина-D1 является относительно универсальным признаком онкогенеза и метастазирования, и его ингибирование или сайленсинг замедляет пролиферацию клеток ГЦР, а также увеличивает их чувствительность к 5-фторурацилу [88]. Ингибиторы PARP (олапариб, нирапариб и др.) в настоящий момент применяются для терапии рака молочной железы и яичников с мутациями BRCA, но рассматриваются также и как альтернативные средства борьбы с ГЦР [89]. Блокада cFLIP позволяет преодолеть приобретенную резистентность клеток к сорафенибу [90] – средству 1-й линии системной терапии ГЦР [4].
В клетках рака предстательной железы SAM вызывал остановку клеточного цикла в S-фазе, снижал экспрессию циклина-А и сигнального белка STAT3, увеличивал активность каспазы-3 и соотношение Bax/Bcl-2, определяющее готовность клетки к апоптозу [91]. Экспериментальный препарат милциклиб, одним из механизмов действия которого является блокада циклина-А, показал высокую эффективность и безопасность у пациентов с нерезектабельной сорафениб-резистентной ГЦР [92], а также в комбинации с регорафенибом – при рецидиве ГЦР после трансплантации печени [93]. Влияние на сигналинг JAK2/STAT3 позволило SAM значительно повысить эффективность гемцитабина в отношении рака поджелудочной железы [66].
SAM синергично с цисплатином снижал экспрессию ряда циклинов, ингибитора циклин-зависимых киназ p21 и угнетал сигналинг Akt, β-катенина и Smad2/3, что вызывало остановку клеточного цикла, замедляло миграцию и инвазию клеток двух линий плоскоклеточного рака головы и шеи, Cal‑33 и JHU‑SCC‑011 [66]. Дисрегуляция сигналинга β-катенина – еще одна характерная черта клеток ГЦР, что делает эту молекулу и ее эффекторы привлекательными мишенями для новых противоопухолевых агентов (малых интерферирующих РНК) [94]. В клетках Cal‑33 и JHU‑SCC‑011 SAM также увеличивал уровни E-кадгерина и снижал – N-кадгерина и виментина, что свидетельствует о его влиянии на процесс эпителиально-мезенхимального перехода клеток [66].
Наконец, SAM продемонстрировал способность регулировать транскрипцию микроРНК-34a, -34c и -486-5p в клетках эстрогензависимого рака молочной железы MCF-7 [95]. МикроРНК семейства 34 являются важным фактором канцерогенеза и чувствительным маркером прогрессии и прогноза при ГЦР [96], а микроРНК-486-5p напрямую подавляет жизнеспособность, пролиферацию, миграцию и клоногенность опухолевых клеток [97].
ВЛИЯНИЕ НА АПОПТОЗ
Отмечается дифференциальный характер влияния SAM на процессы апоптоза: он оказывает проапоптотическое действие на опухолевые, но антиапоптотическое – на нормальные клетки печени. Точные причины такого различия его эффектов не выяснены, однако предполагается, что оно может быть обусловлено различной скоростью конверсии SAM в MTA [98].
SAM частично предупреждал активацию каспазы-3 и ее мишени PARP, опосредующих реакцию апоптоза на сигналы таких факторов, как TGF-β1 и Fas-лиганд, в условиях инкубации нормальных гепатоцитов крысы с окадаевой кислотой. Поскольку MTA, обладавший схожими эффектами, не относится к донорам метильных групп и не участвует в синтезе глутатиона, было выдвинуто предположение, что антиапоптотический эффект этих соединений является самостоятельным и не зависит от антиоксидантной активности [98]. SAM и MTA также стимулировали пролиферацию нормальных гепатоцитов под влиянием HGF. В то же время в клетках ГЦР HuH7, HepG2 и H4-IIE SAM и MTA проявляли синергизм с окадаевой кислотой и оказывали дозозависимое проапоптотическое и антипролиферативное действие [98].
В исследовании in vitro адеметионин уменьшал выраженность характерных признаков апоптоза клеток Saccharomyces cerevisiae, включая конденсацию хроматина, фрагментацию ДНК и перенос фосфатидилсерина на внешний листок плазматической мембраны, вызванных влиянием кислой среды. Основными предполагаемыми механизмами его действия были повышение внутриклеточного уровня глутатиона, замедление перекисного окисления липидов и поддержание потенциала митохондриальных мембран [99].
В отличие от многих антиоксидантов, обладающих антиапоптотическим эффектом, действие SAM, по-видимому, не затрагивает сигналинг c-Jun-N-терминальных киназ (JNK). В первичных гепатоцитах крысы, подвергнутых воздействию токсических концентраций этанола, SAM предупреждал развитие оксидативного стресса, выход цитохрома C в цитоплазму и протеолиз каспазы-3, но не оказывал влияния на активность JNK и фрагментацию проапоптотического белка Bid [100].
ВЛИЯНИЕ НА АУТОФАГИЮ
Последние данные экспериментальных исследований свидетельствуют о роли SAM как сигнальной молекулы, предупреждающей аутофагию, вызванную дефицитом метионина. Путем метилирования ДНК, матричной РНК, гистонов и других белков SAM может ингибировать AMPK-опосредованную аутофагию, а благодаря участию в биосинтезе глутатиона – оказывать влияние и на AMPK-независимые пути [101].
Показана прямая взаимосвязь между SAM и сигнальными путями комплекса-1 мишени рапамицина млекопитающих (mTORC1) посредством специфического сенсора SAMTOR. Высокие концентрации SAM связывают этот белок, тем самым снимая блокаду с комплексов GATOR1 и mTORC1, что, в свою очередь, подавляет процесс аутофагии клетки [102]. В то же время показано, что экзогенный SAM снижает системные уровни гомоцистеина, который служит ингибитором сигналинга mTOR и аутофагии. Спермидин, другой метаболит SAM, также обладает проаутофагическим действием, что указывает на комплексный характер влияния SAM и интермедиатов его метаболических путей на процессы аутофагии [101]. Поскольку дисрегуляция этих процессов является фактором прогрессирования и развития лекарственной устойчивости ГЦР [103], описанные эффекты SAM требуют дальнейшего детального изучения.
Основные механизмы хемопревентивной и противоопухолевой активности SAM при хронических заболеваниях печени приведены на рисунке.
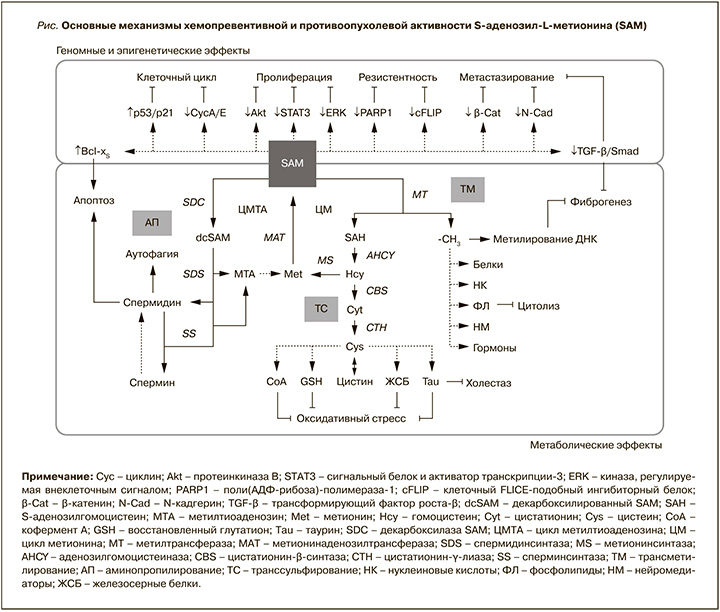
ЗАКЛЮЧЕНИЕ
Агрессивность и устойчивость ГЦР к современным методам лечения определяют исключительную важность его профилактики путем устранения причинных факторов и проведения хемопревенции с помощью фармакологических агентов. Адеметионин (S-аденозил-L-метионин) традиционно известен как интермедиат нескольких биосинтетических путей, обладающий антиоксидантной, антицитолитической и антихолестатической активностью. Одновременно с этим все большее подтверждение находит его участие в регуляции клеточного цикла, процессов пролиферации, эпителиально-мезенхимального перехода, инвазии и метастазирования злокачественных опухолей. Сочетание широкого спектра метаболических и эпигенетических эффектов обусловливает значительный потенциал адеметионина как средства хемопревенции и адъювантной терапии ГЦР.
