
Мукополисахаридозы – группа редких наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов. Они обусловлены мутациями генов, контролирующих процесс внутрилизосомного гидролиза гликозаминогликанов [1–3] и проявляются преимущественно дефектами костной, хрящевой и соединительной тканей [1–5]. Нередко эта группа заболеваний не имеет ярких начальных проявлений, однако в последующем они характеризуются тяжелым течением и неблагоприятным прогнозом, в связи с чем многие пациенты умирают в детском или подростковом возрасте [6].
Единственным существующим в настоящее время и общепринятым патогенетическим методом лечения мукополисахаридозов считается заместительная ферментотерапия [3, 7]. Наиболее заметного эффекта от нее можно добиться при раннем выявлении заболевания и своевременном, регулярном проведении терапии. В связи с этим в литературе последних лет появилось немало работ, посвященных мукополисахаридозам и их диагностике [1, 3, 6–8]. Различают более 10 типов мукополисахаридозов [1, 4], из которых в России наиболее часто встречается синдром Хантера [1, 9].
Синдром Хантера, или мукополисахаридоз II типа, – редкое заболевание с Х-сцепленным рецессивным типом наследования, для которого типично снижение активности лизосомального фермента идуронат-2-сульфатазы из-за мутации в гене IDS [4, 8, 10]. Дефицит этого фермента приводит к аккумуляции гликозаминогликанов, преимущественно гепаран- и дерматансульфатов, в лизосомах практически всех типов клеток различных органов и тканей [9].
Мукополисахаридозом II типа страдают преимущественно мальчики. Частота его составляет 1:140 000–156 000 живорожденных. В то же время описаны единичные случаи синдрома Хантера и у девочек [9].
Клинические проявления синдрома зависят от его формы – легкой, среднетяжелой или тяжелой [1, 4, 9, 10]. Для тяжелой формы характерна манифестация в раннем детском возрасте, для легкой – в 10–15 лет [10]. Заболевание проявляется задержкой роста, выраженными деформациями костей и суставов. Изменения скелета обозначают как «гурлерподобный» фенотип. У больных наблюдаются короткая шея, «когтистая кисть», сгибательные контрактуры тазобедренных, коленных, локтевых суставов, тугоподвижность мелких и крупных суставов, аномалии зубов, макроцефалия, грубые черты лица (большая голова с выступающими лобными буграми, запавшая переносица с широким носом и вывернутыми вперед ноздрями, полные щеки и толстые губы).
Кроме изменений скелета, при синдроме Хантера выявляется патология дыхательной и сердечно-сосудистой систем, гепатоспленомегалия, нарушение слуха, поражения кожи (папулезные «шагреневые» высыпания), атипичный пигментный ретинит, разрушение сетчатки [4, 9, 10, 11]. Тяжелая форма заболевания протекает с вовлечением в патологический процесс нервной системы, в том числе со снижением интеллекта, аномалиями поведения и парезами [1, 4, 9].
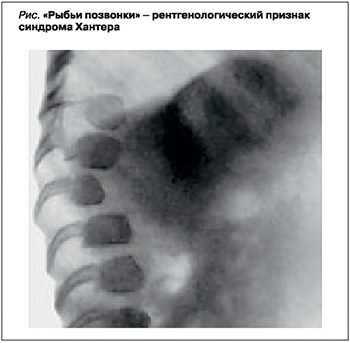 Диагноз «синдром Хантера» ставится на основании клинико-генеалогических данных (отягощенная наследственность, мужской пол, «гурлерподобный» фенотип, постепенно нарастающие изменения) и результатов лабораторных и инструментальных методов исследования [9]. На электрокардиограмме (ЭКГ) выявляются неспецифические изменения – снижение вольтажа желудочковых комплексов QRS. При рентгенологическом исследовании обнаруживаются небольшие сужения проксимальных отделов пястных костей и гипоплазия ногтевых фаланг кистей, признаки раннего окостенения затылочно-теменного шва и так называемые рыбьи позвонки (рис.) [9, 10].
Диагноз «синдром Хантера» ставится на основании клинико-генеалогических данных (отягощенная наследственность, мужской пол, «гурлерподобный» фенотип, постепенно нарастающие изменения) и результатов лабораторных и инструментальных методов исследования [9]. На электрокардиограмме (ЭКГ) выявляются неспецифические изменения – снижение вольтажа желудочковых комплексов QRS. При рентгенологическом исследовании обнаруживаются небольшие сужения проксимальных отделов пястных костей и гипоплазия ногтевых фаланг кистей, признаки раннего окостенения затылочно-теменного шва и так называемые рыбьи позвонки (рис.) [9, 10].
В анализах у таких пациентов отмечается высокая почечная экскреция сульфатированных фракций гликозаминогликанов – гепаран- и дерматансульфатов в моче. При легкой форме болезни тест на гликозаминогликаны может быть ложноотрицательным [4]. Для подтверждения диагноза синдрома Хантера определяется снижение активности фермента идуронатсульфатазы в лейкоцитах, культуре фибробластов кожи, плазме, биоптатах печени и сухом пятне крови [4, 9]. При ДНК-диагностике проводится определение мутации в гене идуронатсульфатазы, в настоящее время описано более 300 мутаций [4]. Обнаружение новых мутаций продолжается по сегодняшний день [12].
В настоящее время в лечении синдрома Хантера применяют внутривенную ферментную заместительную терапию, трансплантацию гемопоэтических стволовых клеток и симптоматическое хирургическое лечение. Трансплантация гемопоэтических стволовых клеток может воздействовать положительно на некоторые неврологические нарушения, но имеет высокий риск посттрансплантационных осложнений [7]. Единственный специфический метод лечения синдрома Хантера на сегодняшний день – ферментозаместительная терапия препаратом идурсульфазы, прием которого способен продлить жизнь больного, а также значительно улучшить его состояние вплоть до остановки прогрессирования заболевания и полной социальной адаптации в обществе [4, 5]. Ограничением внутривенной ферментной заместительной терапии является невозможность фермента проникнуть через гематоэнцефалический барьер (ГЭБ) [8]. Кроме того, ферментозаместительная терапия более эффективна, если ее начинают на более раннем этапе, когда отсутствуют необратимые проявления болезни, что, к сожалению, не всегда возможно [6].
В настоящее время ведутся поиски высокодозной, интратекальной и интравентрикулярной ферментозаместительной терапии с применением модифицированного фермента, способного преодолевать ГЭБ. Также проводятся клинические исследования генной, клеточной, субстратредуцирующей терапии синдрома Хантера и методов считывания через стоп-кодон [13, 14].
Далее мы приводим клиническое наблюдение за больным с этим редким заболеванием.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ
В неврологическое отделение поступил пациент, 19 лет, с жалобами на ограничение движений в плечевых, локтевых, тазобедренных суставах, суставах кистей, судороги в нижних конечностях, онемение и зябкость рук, боли в руках и в ногах, снижение ночного зрения, неуверенность походки, повышенную раздражительность, снижение памяти и внимания.
Из анамнеза известно, что с 6 лет пациента беспокоят боли в коленных суставах, с 7 лет появились ограничения движений в плечевых суставах, с 8 лет – в пальцах рук. В возрасте 6 лет впервые был консультирован генетиком, выявлен нормальный мужской кариотип. В связи с прогрессированием симптоматики в 10 лет пациент был обследован по подозрению на наследственную патологию. Был выполнен одномерный электрофорез гликозаминогликанов, выявлена повышенная экскреция с мочой дерматансульфата и гепарансульфата. По результатам ДНК-анализа гена идуронат-сульфатазы была обнаружена мутация P197L в 5 экзоне гена IDS.
Из перенесенных заболеваний больной отмечал пупочную и двустороннюю паховую грыжи, дискинезию желчевыводящих протоков, пневмонию, отит, удаление аденоидов. Наследственность отягощена: мукополисахаридоз у деда и брата.
Данные физикального обследования: общее состояние больного удовлетворительное. Температура тела 36,7 °С. Макроцефалия. Широкая переносица. Выраженные надбровные дуги. Ограничение движений в локтевых и тазобедренных суставах. Кожные покровы физиологической окраски. В легких дыхание везикулярное, хрипы не выслушиваются. Частота дыхательных движений – 18/ мин. Аускультация сердца: ритм правильный, тоны приглушены. Частота сердечных сокращений – 76 уд./мин. Артериальное давление – 110/70 мм рт.ст. Язык влажный, чистый. Живот мягкий, безболезненный. Симптом поколачивания отрицательный с обеих сторон. Сознание ясное. Лицо симметричное. Язык по средней линии. Нистагма нет. Менингеальных знаков нет. Чувствительных нарушений нет. В пробе Ромберга устойчив. Объем активных и пассивных движений в суставах ограничен из-за контрактур. Гипергидроз ладоней и стоп. Формирующиеся «когтистые» кисти и стопы.
В общеклинических и биохимических анализах показатели в пределах нормы.
Пациенту проведено симптоматическое лечение (витамины, сосудистые препараты). Однако прогноз остается неблагоприятным, так как с возрастом будут нарастать изменения скелета, нарушения функций различных органов и систем.
ОБСУЖДЕНИЕ
Современные достижения медицины позволяют продлить жизнь и улучшить ее качество пациентам со многими заболеваниями, в том числе с синдромом Хантера. Они могут доживать до 50 лет, обучаться, успешно заканчивать высшие учебные заведения, работать, вступать в брак и иметь здоровое потомство. Такие результаты возможны прежде всего при условии раннего начала ферментозаместительной терапии. В связи с этим спектр специалистов, к которым они могут обращаться за помощью, расширяется. Необходимо иметь настороженность в отношении наследственной патологии у детей с частыми респираторными заболеваниями, отставанием в развитии и изменениями скелета. В настоящее время не все врачи помнят о данной патологии, поскольку она относится к редким заболеваниям. В то же время раннее проведение ДНК-диагностики позволяет своевременно начать ферментозаместительную терапию и предотвратить инвалидизацию больного в детском и подростковом возрасте. Надеемся, что представленный клинический случай будет иметь большой практический интерес не только для педиатров и генетиков, но и для врачей других специальностей.


