
Лонг-ковид и постковидный синдром: определения
До настоящего времени мировое научное сообщество так и не выработало единых подходов к определению терминов «лонг-ковид» и «постковидный синдром», хотя эти понятия появились более года назад, спустя несколько месяцев после начала пандемии. Всемирная организация здравоохранения (ВОЗ) также пока находится на этапе представления протокола, который разработан для продвижения в вопросах, касающихся формулировки определения постковидного синдрома [1]. При этом поток пациентов, обращающихся с долговременными последствиями COVID-19 к разным специалистам, становится все больше, и российское медицинское сообщество нуждается хотя бы во временных, но четких определениях терминов, а также рекомендациях по ведению таких пациентов.
Согласно многим публикациям, под термином «лонг-ковид» принято понимать клинические проявления заболевания, длящиеся более 4, но менее 12 нед с начала болезни, а под термином «хронический ковид», или «постковидный синдром», – период, выходящий за рамки 12 нед с момента развития заболевания (рис.) [2–5].
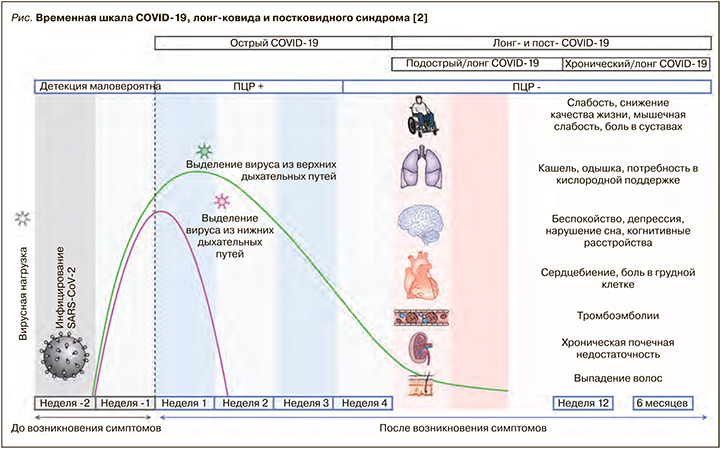
Многие исследователи склонны считать, что положительный тест на COVID-19 (в качестве лабораторного подтверждения диагноза) не является обязательным условием для постановки «лонг-ковида» и постковидного синдрома, так как нередко встречаются ложноотрицательные результаты [6, 7].
Предполагается, что вирус, отвечающий за репликацию, исчезает максимум через месяц после возникновения симптомов, оставляя после себя долговременные последствия [2]. Тем не менее сейчас многие исследования сосредоточены на поиске резервуаров вируса, в которых он предположительно может сохраняться в течение длительного времени. Это не означает, что вирус в резервуаре обязательно способен к репликации, но длительное сохранение мРНК или ее фрагментов в клетках организма человека может способствовать развитию хронического воспалительного процесса и дисрегуляции иммунной системы. В частности, даже через несколько месяцев после заражения в биоптатах кишечника людей, перенесших COVID-19, обнаружена мРНК вируса SARS-CoV-2, а также вирусный белок, на который может реагировать иммунная система [8].
Тем не менее в настоящее время для больных с лонг-ковидом и постковидным синдромом использование противовирусных препаратов не рекомендовано. Несколько особняком стоят лишь пациенты, у которых на протяжении нескольких месяцев сохраняется положительный результат полимеразной цепной реакции (ПЦР) на SARS-CoV-2 в назофарингеальных мазках. Как правило, это пациенты с онкологической патологией, заболеваниями системы крови, ВИЧ-инфекцией [9]. У данной группы пациентов может быть эффективным использование иммунотерапии для достижения клиренса вируса [10], хотя применение противовирусных средств у них также не приносит ожидаемого эффекта [11].
Патофизиологические процессы, лежащие в основе лонг-ковида и постковидного синдрома
Выделяют несколько механизмов, оказывающих влияние на развитие лонг-ковида и постковидного синдрома: иммунная дисрегуляция и аутоиммунные механизмы, синдром системного воспалительного ответа, нарушение гемостаза и васкулопатия; прямое цитотоксическое действие вируса на клетки и длительная вирусная персистенция; вегетативная, нервная, эндокринная и метаболическая дисфункция; дисбаланс в функционировании пептидов, образующихся в результате действия АПФ1- и АПФ2-рецепторов, а также последствия состояния пациента, находившегося в критическом состоянии.
Совокупное и в разной степени выраженное влияние этих механизмов и приводит к разнообразным клиническим проявлениям, отмечающимся у пациентов после перенесенного COVID-19.
• Патофизиология синдрома, связанного с последствиями состояния пациента, нуждавшегося в интенсивной терапии, является многофакторной и включает повреждение сосудов микроциркуляторного русла, метаболические изменения, связанные с критическим состоянием и неподвижностью [12]. Кроме этого, выжившие после COVID-19 пациенты могут подвергаться повышенному риску вторичных инфекций, вызванных бактериальными, грибковыми (аспергиллез легких) или другими патогенами [13].
• Повреждение респираторного тракта обусловлено вирусозависимыми (включая поражение вирусом SARS-CoV-2 клеток альвеолярного эпителия и эндотелиальных клеток) и вирусонезависимыми механизмами (такими как иммунологическое повреждение, в том числе периваскулярное воспаление), которые совместно способствуют разрушению эндотелиально-эпителиального барьера и экстравазации богатого белками экссудата в альвеолярное пространство [14].
Диффузное повреждение альвеол с последующей организацией и очаговым фибропролиферативным диффузным поражением ткани легких наблюдается в поздние сроки COVID-19 [15]. Нередко отмечаются очаги пролиферации миофибробластов и фиброзно-кистозного перерождения легких. Этот процесс может быть спровоцирован провоспалительными цитокинами (интерлейкином-6 и трансформирующим фактором роста-β) [16] и в дальнейшем предрасполагать к бактериальной колонизации и последующей вторичной инфекции [17]. Свой вклад в поражение легких при лонг-ковиде и постковидном синдроме вносят также последствия микро- и макротромбоза легочных сосудов, эндотелиальная дисфункция и тромботическая микроангиопатия, наблюдающиеся у 20–30% пациентов с COVID-19 на фоне гипоксии, гипервоспаления и гиперкоагуляции, активации комплемента, тромбоцитов, взаимодействия тромбоцитов с лейкоцитами, высвобождения провоспалительных цитокинов [18, 19]. Риск тромботических осложнений в период лонг-ковида и постковидного синдрома, вероятно, обусловлен продолжительностью и тяжестью гипервоспалительного состояния [2].
• К механизмам, определяющим сердечно-сосудистые осложнения при лонг-ковиде и постковидном синдроме, относят прямое воздействие вируса на клетки, снижение уровня активных АПФ2-рецепторов, воспаление и иммунологический ответ, влияющий на структурную целостность миокарда, перикарда и проводящей системы сердца, гибель кардиомиоцитов и фиброзно-жировые замещение десмосомных белков, играющих важную роль в межклеточном сцеплении [20].
Выздоровевшие пациенты могут испытывать устойчиво повышенную потребность в препаратах, улучшающих метаболизм миокарда. Это может быть связано со снижением сердечного резерва, применением кортикостероидов и нарушением регуляции ренин-ангиотензин-альдостероновой системы (РААС). Фиброз или рубцевание миокарда и возникающая в результате этого кардиомиопатия могут привести к суправентрикулярной тахиаритмии [21].
COVID-19 может также способствовать развитию аритмии из-за повышенного катехоламинергического состояния, связанного с провоспалительными цитокинами – интерлейкином-6 (ИЛ-6), ИЛ-1 и фактором некроза опухоли-альфа (ФНО-α), которые способны продлевать потенциалы действия желудочков путем модуляции экспрессии ионных каналов кардиомиоцитов [22]. Кроме этого, любая вирусная инфекция сама по себе приводит к синдрому постуральной ортостатической тахикардии и синусовой тахикардии в результате адренергической модуляции [23].
• Механизмы, способствующие развитию патологии нервной системы при COVID-19, могут быть также объяснены непосредственным воздействием вируса и нейровоспалением, тяжелым системным воспалением, микрососудистым тромбозом и нейродегенерацией [24–26]. Пока нет убедительных доказательств инфицирования нейронов SARS-CoV-2, однако этот вирус может вызывать изменения в паренхиме головного мозга и сосудах, по-видимому, воздействуя через ГЭБ и вызывая воспаление в нейронах, васкуляризирующихся сосудами головного мозга [27, 28].
Помимо этого, уровни иммунной активации напрямую коррелируют с когнитивно-поведенческими изменениями. Хроническое вялотекущее воспаление головного мозга, наряду со сниженной способностью реагировать на новые антигены и накоплением Т-клеток памяти (признаки иммуносенсибилизации при старении и повреждении тканей), может играть роль в развитии стойких симптомов COVID-19 [29]. Другие предполагаемые механизмы включают дисфункциональный лимфодренаж из периваскулярного пространства, а также проникновение вирусов во внеклеточные пространства обонятельного эпителия, их пассивную диффузию и аксональный транспорт через обонятельный тракт [30, 31].
Биомаркеры повреждения головного мозга, такие как повышенные уровни легкой цепи нейрофиламентов в периферической крови, были обнаружены у пациентов с COVID-19 с более тяжелой и длительной инфекцией [32, 33], что предполагает возможность хронического повреждения нейронов.
Постковидный «мозговой туман» у тяжелобольных пациентов с COVID-19 может быть связан с декондиционированием или посттравматическим стрессовым расстройством [34]. Однако сообщения о «мозговом тумане» после легкой формы болезни предполагают, что дисфункция вегетативной нервной системы также может влиять на его развитие [35, 36]. Отметим, что долгосрочные когнитивные нарушения после перенесенного критического состояния встречаются у 20–40% пациентов [37].
• Эндокринные проявления после перенесенного COVID-19 могут быть последствиями прямого повреждения вирусом SARS-CoV-2, дисрегуляции иммунной системы и воспаления, ятрогенных осложнений. Ранее существовавший диабет может впервые манифестировать в острой фазе COVID-19 и, как правило, лечится безинсулиновыми препаратами даже при изначальном развитии диабетического кетоацидоза. Нет конкретных доказательств длительного повреждения β-клеток поджелудочной железы при COVID-19 [38].
Есть данные, что экспрессия АПФ2 и трансмембранной сериновой протеазы (TMPRSS2), участвующей в проникновении SARS-CoV-2 в β-клетки, и первичный дефицит инсулина, наряду с резистентностью к инсулину, вероятно, опосредованы такими факторами, как воспаление или реакция на инфекционный стресс [38]. До сих пор не найдено подтверждений того, что диабет, связанный с новой коронавирусной инфекцией, может регрессировать после острого COVID-19, а также того, что его течение при лонг-ковиде и постковидном синдроме чем-то отличается от течения диабета у больных, не болевших COVID-19.
COVID-19 также повышает риски деминерализации костей, связанные с системным воспалением, иммобилизацией, воздействием кортикостероидов, дефицитом витамина D и прекращением приема антирезорбтивных или анаболических средств при остеопорозе в острый период болезни [39].
• Патология почек. SARS-CoV-2 был выделен из почечной ткани [40], и острый некроз канальцев является основной находкой, отмеченной при биопсиях почки [41]. COVID-19-ассоциированная нефропатия характеризуется коллапсирующим вариантом очагового сегментарного гломерулосклероза с инволюцией клубочкового аппарата в дополнение к острому повреждению канальцев и, как полагают, развивается в ответ на активацию интерферона и хемокинов [42]. Ассоциация с аллелями риска APOL1 предполагает, что SARS-CoV-2 действует как триггер у предрасположенных к этой патологии пациентов [2]. Тромбы в сосудах микроциркуляторного русла почек также могут потенциально способствовать их повреждению [43].
• Патология желудочно-кишечного тракта (ЖКТ). COVID-19 обладает потенциалом для воздействия на микробиом кишечника, включая обогащение условно-патогенными микроорганизмами и истощение полезных комменсалов [44, 45]. Способность микробиоты кишечника изменять течение респираторных инфекций (ось «кишечник–легкие») была признана ранее при гриппе и других респираторных инфекциях [46]. В настоящее время проводятся исследования по оценке долгосрочных последствий COVID-19 для ЖКТ, включая синдром раздраженного кишечника и диспепсию [2].
• Мультисистемный воспалительный синдром. Заболеваемость COVID-19 в эпицентрах весной 2020 г. и обнаружение после весеннего пика с интервалом примерно через месяц проявлений мультисистемного воспалительного синдрома у пациентов с уже отрицательным результатом ПЦР, но положительными тестами на антитела позволяют предполагать, что этот синдром – результат искаженной реакции приобретенного иммунитета, а не острой вирусной инфекции [47]. Понимание патофизиологии мультисистемного воспалительного синдрома может быть частично получено исходя из патогенеза болезни Кавасаки и синдрома токсического шока; она объясняется возможными механизмами повреждения, обусловленными иммунными комплексами, активацией комплемента, образованием аутоантител и массивным высвобождением цитокинов, связанных со стимуляцией суперантигенами Т-клеток [48, 49].
Таким образом, патологическое воздействие SARS-CoV-2 на организм человека многогранно и отражается на его функционировании даже после перенесенного острого COVID-19. У одних пациентов проявления лонг-ковида и постковидного синдрома практически не выражены, у других, даже перенесших инфекцию в легкой форме, отмечаются долговременные последствия, влияющие на трудоспособность и качество жизни. Подход к лечению лонг-ковида и постковидного синдрома должен включать комплексные методы воздействия на организм, ориентированные на снижение выраженности или устранение органной дисфункции, и реабилитационные мероприятия, основанные на понимании патофизиологии этих состояний.


