
Дилатационная кардиомиопатия (ДКМП) — наиболее часто встречающееся среди первичных поражений миокарда заболевание, характеризующееся высокой смертностью. По данным Европейского общества кардиологов, летальность при ДКМП составляет 20% в течение 5 лет после установления диагноза. Распространенность заболевания в европейской популяции составляет более 37 случаев на 100 тыс. населения [1].
Основными причинами смерти при ДКМП являются внезапная сердечная смерть (ВСС) или смерть от декомпенсированной застойной сердечной недостаточности (СН). ДКМП может быть результатом вирусных миокардитов, системных заболеваний, токсических воздействий, метаболических и аутоиммунных расстройств, а также генетических нарушений. Заболевание отличается выраженной клинической и генетической гетерогенностью [1—3].
В настоящее время известно более 50 генов, мутации которых ассоциированы с ДКМП [4—7]. По данным разных авторов, на долю первичных ДКМП, обусловленных мутациями генов, которые кодируют структурные белки миокарда, приходится не менее 25—40% зарегистрированных случаев, по некоторым данным — до 60% [5—8].
В последнее десятилетие активно изучается ламиновый генотип ДКМП, прогностически наиболее неблагоприятный среди других генных мутаций.
Впервые мутации в гене ламина (LMNA), вызывающие развитие ДКМП с нарушением проводимости, выявлены D. Fatkin в 1999 г., затем это открытие инициировало целый ряд аналогичных исследований [9—12]. Согласно результатам мета-анализа, проведенного Van Berlo и C. Meune, у пациентов — носителей ламиновых генных аномалий, отмечается высокая летальность: в течение 3-летнего периода наблюдения зарегистрировано 46% случаев ВСС, несмотря на наличие пейсмейкеров в группе больных брадиаритмиями [11]. В исследовании М. Pasotti период наблюдения пациентов с ламин-ассоциированной ДКМП составил 36—107 (медиана 57) мес: в 55,1% случаев выявлены желудочковые тахиаритмии, в 24,5% имплантированы кардиовертерыдефибрилляторы (КД), в 32,7% зарегистрирована ВСС, а в 30,6% случаев проведена трансплантация сердца [12]. В период с 2009 по 2012 г. европейскими кардиологами (рабочие группы специалистов по проблеме) и Американской ассоциацией по сердечной недостаточности обновлены практические рекомендации по генетической диагностике кардиомиопатий (КМП) с включением специальной позиции по лечению и диагностике ламин-связанной ДКМП [13—15]. Основные положения европейских и американских кардиологов совпадают по концепции выделения ламиновых фенотипов ДКМП для обязательного молекулярно-генетического тестирования и в случае идентификации LMNA-ассоциированной ДКМП – ранней имплантации КД для профилактики ВСС [16—18].
Ламин A/C — структурный белок ядерной ламины, относящийся к классу промежуточных филаментов V. Ядерная ламина — это структурный элемент ядра, который противостоит силам деформации и защищает хроматин от физических повреждений. Ламина предопределяет размер, форму и прочность ядерной оболочки. Свойства этого элемента зависят не только от структурного строения ламинов, но и от способа его взаимодействия с компонентами цитоскелета клетки [19]. С помощью основного домена ламины формируют димер и взаимодействуют с хроматином и другими ключевыми белками внутренней ядерной мембраны, ламиновые комплексы принимают участие и в регуляции транскрипционных процессов. Снижение (потеря) функции, в частности ламина A, ведет к дегенерации, снижению нормальной регенерации клеток в различных тканях и к преждевременной клеточной гибели — апоптозу [20].
Мутации гена LMNA служат причиной 9 различных наследственных заболеваний, получивших название «ламинопатии» (в том числе мышечная дистрофия Эмери—Дрейфуса, поясно-конечностная мышечная дистрофия 1В типа, ДКМП 1А типа, болезнь Шарко—Мари—Тута типа 2В1, семейная липодистрофия Даннигана, акромандибулярная дисплазия, синдром прогерии Хатчинсон—Гилфорда). Ламинопатии отличаются значительной фенотипической и генетической гетерогенностью.
Они являются результатом миссенс-мутаций (замена аминокислоты в белке) в кодирующих частях гена ламина (72% всех изученных мутаций LMNA), в сайтах сплайсинга на границе экзон—интрон (7%), а также вставок/делеций нуклеотидов в кодирующих (9%) и некодирующих (9%) частях гена. Встречаются также нонсенс-мутации (5%), приводящие к преждевременному обрыву синтеза цепи белка — «стоп кодон» [21—24]. Практически все идентифицированные в гене LMNA мутации зарегистрированы и доступны для ознакомления в европейской базе данных UMD-LMNA мутаций (www.umd.be/LMNA) и американской базе данных (www.hgvs.org; Human Intermediate Filament Database LMNA; IPN Mutations LMNF; LMNA homepage — Leiden Muscular Dystrophy pages).
Наряду с дилатацией и снижением систолической функции левого желудочка (ЛЖ) ламиновый фенотип почти всегда сопровождается клинически значимыми нарушениями проводимости и ритма [10—13, 16—18]. Болезнь может дебютировать (и в раннем возрасте, и в 3—4-й декадах жизни) с манифестации дефектов проводящей системы (чаще атриовентрикулярные – АВ-блокады и клинически проявляющиеся брадиаритмии, в том числе синдром слабости синусного узла) или аритмии (наджелудочковые тахиаритмии — фибрилляция/трепетание предсердий; желудочковые тахиаритмии — тахикардия/фибрилляция/трепетание желудочков). Нередко синкопальные состояния являются первыми и последними клиническими проявлениями заболевания, отмечаются случаи ВСС даже при незначительной (или в ее отсутствие) систолической дисфункции ЛЖ [24—31]. КМП, вызванные мутациями LMNA, часто сопровождаются скелетно-мышечными расстройствами (периферические миопатии) различной степени выраженности. Чаще встречается мышечная дистрофия Эмери—Дрейфуса, наследуемая по аутосомно-доминантному типу, но может – и по аутосомно-рецессивному типу, и поясно-конечностная мышечная дистрофия с нарушением АВ-проводимости типа 1В. Иногда отмечаются изолированные скелетно-мышечные гипо/гипертрофии конечностей [21—24].
Мы представляем клинический случай смешанного фенотипа ДКМП, ассоциированной с новой мутацией гена LMNA, тяжелыми нарушениями ритма сердца и проводимости, бивентрикулярной дисфункцией и незначительными изменениями скелетных мышц нижних конечностей.
Клинический случай
Пациентка К., 23 года, без семейного анамнеза ДКМП, наблюдалась до 16 лет у педиатра по поводу вегетососудистых и миастенических жалоб (предполагалась поясно-конечностная миопатия, но диагноз не был верифицирован из-за отказа родителей от обследования). В подростковом возрасте клинически значимых нарушений ритма сердца и проводимости не отмечалось; по результатам суточного мониторирования электрокардиограммы (ЭКГ) была выявлена одиночная предсердная экстрасистолия и интермиттирующая АВ-блокада I степени; по данным эхокардиографии (ЭхоКГ), структурно-функциональных аномалий (дисфункция миокарда, изменения размеров камер) сердца также не отмечалось. В последующие 5 лет пациентка за медицинской помощью не обращалась.
Первые жалобы на сердцебиение и нарушения ритма сердца появились в 21-летнем возрасте. На ЭКГ выявлены низкий вольтаж комплексов QRS в стандартных и левых грудных отведениях, электрическая ось сердца типа QI — QII — QIII, плохо визуализируемый низкоамплитудный зубец Р, синусовая брадикардия с частотой сердечных сокращений (ЧСС) 50 уд/мин, АВ-блокада I степени, одиночные предсердные экстрасистолы, в том числе с блокированным проведением. При ЭхоКГ видимой патологии не обнаружено: конечный диастолический диаметр (КДД) ЛЖ 48 мм, конечный систолический диаметр (КСД) ЛЖ 29 мм, конечный диастолический объем (КДО) ЛЖ 123 мл, конечный систолический объем (КСО) ЛЖ 47 мл; КДО правого желудочка (ПЖ) 63 мл, КСО ПЖ 28 мл, фракция выброса (ФВ) ЛЖ 62% (по Симпсону), ФВ ПЖ 55%. При холтеровском мониторировании (ХМ) ЭКГ выявлены признаки бинодальной дисфункции: синусовая брадикардия (средняя ЧСС 53 уд/мин, максимальная ЧСС 89 уд/мин, минимальная ЧСС 33 уд/мин); интермиттирующая АВ-блокада I и II степени (Мобитц 1—2); патологическое число (40 в час) наджелудочковых экстрасистол с постэктопическим угнетением функции синусного узла (максимальные паузы до 2,1 с); блокированные (в том числе парные и групповые) предсердные экстрасистолы с паузами до 2,7—2,8 с; пароксизмы неустойчивой фибрилляции/трепетания предсердий от 2 до 15 с.
Субъективно синдром бинодальной слабости был бессимптомным (отсутствие одышки, головокружения и синкопе), и молодая женщина отказалась от имплантации электрокардиостимулятора.
Через 2 года пациентка госпитализирована в республиканский кардиологический центр с клиническими признаками СН, соответствующими III—IV функциональному классу (ФК) по классификации Нью-Йоркской ассоциации сердца (NYHA), с жалобами на одышку, приступы удушья по ночам, тяжесть и боли в правой подреберной области, синкопе и предобморочные состояния. Клиническая картинаа СН развивалась быстро — в течение 3 мес с момента появления первых симптомов. Со слов пациентки, обмороки появились ранее — в течение последнего года (4 синкопе), а одышка при ходьбе и наклонах туловища вперед («bendopnea») — 2—3 мес назад.
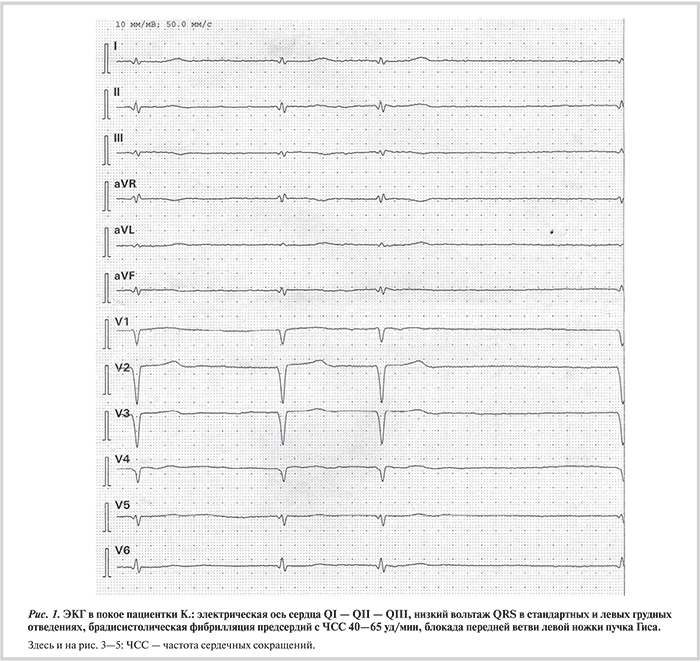
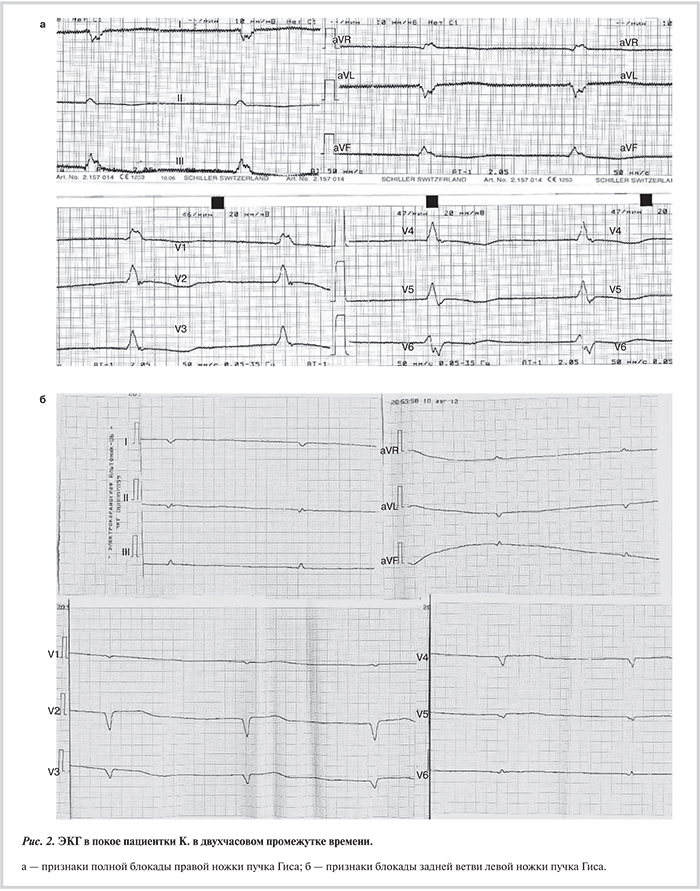
Объективно при осмотре: одышка в покое — частота дыхательных движений 21 в 1 мин, легкий акроцианоз, пульс 51, ЧCC 60 уд/мин, артериальное давление 100/60 мм рт.ст., тоны сердца аритмичные; перкуторно — расширение границ сердца вправо, разлитой верхушечный толчок; аускультативно — систолический шум над мечевидным отростком, мелкопузырчатые хрипы в нижних отделах легких; пальпаторно — увеличение правой доли печени на 2,5 см от края реберной дуги; периферические отеки стоп и голеней, признаки гипертрофии икроножных мышц, умеренно выраженная гипотрофия бедренных мышц с незначительным снижением мышечной силы и без аномалии периостальных рефлексов. На ЭКГ: фибрилляция предсердий (ФП) с ЧСС 51—60 уд/мин, неполная блокада левой ножки пучка Гиса (ЛНПГ) — альтернирующие блокады передневерхнего и задненижнего разветвления, интермиттирующая полная блокада правой ножки пучка Гиса (БПНПГ) (рис. 1, 2). При ЭхоКГ выявлены дилатация и глобальная систолическая дисфункция обоих желудочков с дилатацией предсердий: ФВ ЛЖ 27%, продольная деформация ЛЖ (mean global strain — GS) — 8,6%; ФВ ПЖ 39%, продольная деформация ПЖ (GS) —9,8%; КДД ЛЖ 56 мм (индекс 28 мм/м2), КСД ЛЖ 46 мм, КДО ЛЖ 171 мл, КСО ЛЖ 124 мл; КДО ПЖ 122 мл, КСО ПЖ 74 мл, конечный диастолический размер выходного тракта (ВТ) ПЖ по длинной оси 40 мм (индекс 24 мм/м2); недостаточность митрального и трикуспидального клапанов c митральной регургитацией II степени и трикуспидальной регургитацией III степени; объем левого предсердия 78 мл, объем правого предсердия 200 мл; среднее давление в легочной артерии (срДЛА) 28 мм рт.ст. Обнаружены признаки трабекулярного строения верхушки ЛЖ, заднебоковых отделов ЛЖ, верхушки ПЖ, межжелудочковой перегородки со стороны ПЖ, выявлен некомпактный миокард свободной стенки ПЖ. Толщина миокарда ПЖ 7 мм, толщина миокарда ЛЖ 10 мм.
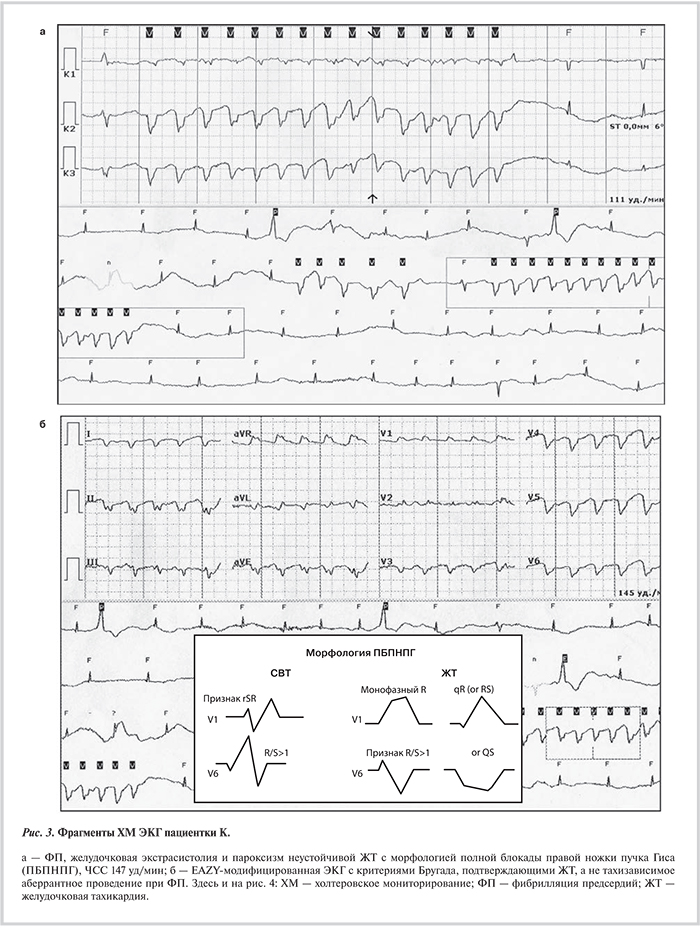
С помощью кардиореспираторного теста определен максимальный объем потребления кислорода (VO2) в последние 30 с физической нагрузки, пик VO2 составил 15,5 мл/кг/мин (диапазон половозрастной нормы 29—42 мл/кг/мин). При ХМ ЭКГ выявлены следующие нарушения ритма и проводимости сердца: ФП брадисистолической формы со средней ЧСС 45 уд/мин; частая полиморфная желудочковая экстрасистолия (>300/ч), в том числе парная (345 куплетов/сут) и групповая (13 триплетов/сут); в ночное и утреннее время зарегистрированы альтернирующие эпизоды синдрома Фредерика (АВ-блокада III степени дистального типа общей длительностью 15 ч/сут с минимальной ЧСС 24 уд/мин) и 40 пароксизмов неустойчивой желудочковой тахикардии (ЖТ) двух морфологий (паттерны в виде БПНПГ и задненижнего разветвления ЛНПГ). Фрагменты ХМ ЭКГ представлены на рис. 3, 4.

По данным анамнеза, объективных, лабораторных, инструментальных методов исследования были исключены экстракардиальные причины ФП: метаболические и электролитные нарушения, заболевания легких, интоксикации и патология щитовидной железы (уровни гормонов Т3, Т4, тиреотропного гормона и антитела к нему в норме).
При лабораторном биохимическом исследовании крови выявлено повышение уровней сывороточной креатинфосфокиназы (КФК) — 293 ед/л (норма 24—190 ед/л) и натрийуретического пептида — 789 пг/ммоль (норма 0—50 пг/ммоль). При вирусологическом (метод полимеразной цепной реакции) исследовании генетические маркеры (РНК, ДНК) 8 кардиотропных вирусов (энтеровирус, вирус Эпштейна—Барр, вирус ветряной оспы, парвовирус В19, аденовирус, вирус простого герпеса, вирус герпеса человека 6-го типа и цитомегаловирус) в сыворотке и клеточных элементах крови не обнаружены, серологические тесты (титры антител к антигенам кардиомиоцитов и вирусов) были отрицательными.
При селективной ангиографии коронарных артерий видимых патологических изменений коронарного русла не обнаружено.
С помощью сцинтиграфии миокарда (в состоянии покоя) выявлены зоны гипоперфузии в передней, переднебоковой, передневерхушечной и задней областях ЛЖ (рис. 5, см. цв. вклейку).
Магнитно-резонансное исследование (МРИ) выполнено пациентке на абдоминальной катушке в неполном объеме из-за значительных клинических проявлений левожелудочковой СН в горизонтальном положении в условиях сканирования. На полученных срезах выявлены диффузная дилатация всех камер сердца (в большей степени правых отделов); расширение легочного ствола до 44 мм (превышение диаметра аорты); снижение глобальной сократимости миокарда ЛЖ и ПЖ; локальные мелкие аневризматические выпячивания свободной стенки ПЖ без признаков истончения, без жировой инфильтрации миокарда и эпикарда; выраженная трабекулярность миокарда ПЖ (отношение некомпактной к компактной части миокарда составило более 2:1); жидкость в полости перикарда и в видимых отделах брюшной полости. Заключение: МР-признаки ДКМП, нельзя исключить аритмогенную правожелудочковую кардиомиопатию (АПЖК)/дисплазию ПЖ.
С согласия пациентки ее биологический материал был направлен на молекулярно-генетическое исследование для поиска мутаций в гене ламина. Еще до подтверждения информации о ламиновом генотипе пациентке проведена имплантация однокамерного КД.
Несмотря на проведение адекватной медикаментозной терапии (лечение СН ингибиторами ангиотензинпревращающего фермента, β-адреноблокаторами, антагонистами альдостерона, диуретиками), наблюдалось дальнейшее прогрессирование симптомов СН с развитием асцита и гидроперикарда. При контрольном ЭхоКГ через 2,5 мес после имплантации КД выявлены признаки прогрессирующего негативного ремоделирования сердца: увеличение КДО ЛЖ до 180 мл и КСО ЛЖ до 140 мл; снижение ФВ ЛЖ до 22% и срGS ЛЖ до -5,2%; увеличение КСО ПЖ до 93 мл и КДО ПЖ до 131 мл; уменьшение ФВ ПЖ до 29% и GS ПЖ до -4,9%; появление признаков выраженной легочной гипертензии (срДЛА 46 мм рт.ст.).
Пациентку включили в лист ожидания трансплантации сердца и через 90 дней после имплантации КД проведена успешная ортотопическая трансплантация сердца.

В гистологических образцах биоптатов миокарда эксплантированного сердца выявлены неспецифические изменения, характерные для ДКМП: диффузные изменения миоцитов (включая вариации размера миоцитов и ядер), интерстициальный фиброз, жировая инфильтрация. При морфометрии в двух образцах из отделов ПЖ выявлено фиброзно-жировое замещение до 40% кардиомиоцитов.
Генетическое типирование. Молекулярно-генетическоое исследование заключалось в поиске мутаций в гене LMNA методами анализа однонитевого конформационного полиморфизма (SSCP) и прямого секвенирования. По результатам анализа было выявлено гетерозиготное носительство мутации Arg190Pro в 3-м экзоне. Согласно базам данных SNP, эта замена соответствует rs267607571, в которой ранее были описаны другие мутации — Arg190Gln, Arg190Trp, Arg190fsX22, связанные с ДКМП [32].
Тяжесть проявления идентифицированной нами мутации, возможно, связана с особенностями пролина, включенного в так называемый rod-домен ламина А/С. Этот домен непосредственно участвует в димеризации ламинов А/С с образованием суперспиральной структуры (coiled-coil). Следует отметить, что все аминокислоты характеризуются разной способностью образовывать α-спирали, среди них пролин обладает крайне низким потенциалом к формированию α-закрученных структур [33]. В связи с этим его включение в положение 190 аминокислотной цепочки белка изогнет α-спираль на 30° от основной оси. Такие молекулярные изменения во вторичной структуре ламина А/С неизбежно приведут к нарушению процесса димеризации, а в дальнейшем и полимеризации этого белка в единую сеть. Изменение вторичной структуры ламина А/С и вероятность образования суперспиралей rod-доменами были проанализированы с помощью специальных алгоритмов Jpred3 и NetSurfP [34, 35].
По результатам анализа оказалось, что мутация Arg190Pro с большой вероятностью будет препятствовать образованию суперспиральной структуры. Такие молекулярные изменения в структуре ламина А/С могут снижать механическую стабильность миоцитов, что является критичным во время мышечного, в том числе кардиального, сокращения.
Обсуждение
Представленный клинический случай демонстрирует весьма актуальную проблему трансформирующихся, «смешанных», «полиморфных» и «перекрывающихся» фенотипов КМП и связанные с этим сложности в диагностике и лечении таких заболеваний. Пациентка до 16 лет являлась бессимптомным носителем ламиновой мутации, затем заболевание дебютировало с нарушения АВ-проводимости I степени, а к 21-му году жизни выявлены признаки бессимптомной бинодальной дисфункции без видимой морфофункциональной патологии сердца. В 23-летнем возрасте стремительное развитие КМП с бивентрикулярной дисфункцией и прогрессированием СН за короткий период времени (3—6 мес) ускорило проведение ургентной трансплантации сердца пациентке.
Представленный клинический случай сложно классифицировать по нескольким причинам. Первая — это парадигма единого клинического фенотипа. Наличие морфофункционально смешанных и «перекрывающихся» форм КМП не предусмотрено ни в одной из существующих нозологических классификаций. В представленном случае наблюдается ламиновый фенотип ДКМП, бивентрикулярный подтип АПЖК с вовлечением ЛЖ и сомнительная форма некомпактного строения миокарда ЛЖ. Описанный случай демонстрирует все клинические признаки ламин-ассоциированной ДКМП: 1) критерии L. Mestroni [36, 37]: ФВ ЛЖ 27% (<45%); фракция укорочения 20% (<25%); КДД ЛЖ 56 мм и индекс КДД ЛЖ 28 мм/м2 (корригированный по возрасту и площади поверхности тела индекс КДД>117%); 2) манифестация болезни, предшествующая дилатации и дисфункции сердца, в виде дефектов проводимости и нарушения ритма: интермиттирующая АВ-блокада I и II степени (Мобитц 1—2), наджелудочковая экстрасистолия, хронотропная дисфункция синусного узла, пароксизмы неустойчивой ФП; 3) скелетная миопатия, часто сопутствующая ламин-связанной ДКМП (гипотрофия мышц нижних конечностей и повышение уровня сывороточной КФК); 4) брадиаритмии с прогрессирующим нарушением АВ-проводимости до полной поперечной блокады сердца, появление синкопе и неустойчивой ЖТ также характерны для ДКМП 1А и 1Б типов [38].

Однако на основании модифицированных европейских критериев (F.I. Marcus, W.J. McKenna, 2010 г) выявлены 1 «большой» и 2 «малых» критерия, позволяющие верифицировать диагноз дисплазии ПЖ [39]. По данным ЭхоКГ, подтвержденным результатами МРИ, выявлен «большой» признак АПЖК: КДР ВТ ПЖ (длинная ось) 40 мм с индексом 24 мм/м2 (критерий — ПЖ в конце диастолы ≥32 мм, индекс ≥19 мм/м2) и локальные мелкие аневризматические выпячивания свободной стенки ПЖ. По данным ХМ ЭКГ и результатам гистологической морфометрии определены 2 «малых» признака: 1) частая полиморфная желудочковая экстрасистолия >300 в час (критерий >500 правожелудочковых экстрасистол в сутки) и пароксизмы неустойчивой ЖТ с морфологией полной БПНПГ и блокады задненижнего разветвления ЛНПГ (критерий — пароксизмы ЖТ с морфологией блокады ЛНПГ); 2) наличие ≥60% остаточного количества кардиомиоцитов с 40% фиброзно-жировым замещением миокарда свободной стенки ПЖ (по результатам морфометрии в двух образцах эксплантированного сердца). При применении критериев W.J. McKenna (1994) наличия двух «больших» признаков (выраженная дилатация ПЖ со снижением ФВ ПЖ и локальными аневризмами ПЖ, фиброзно-жировое замещение миокарда по данным микроскопии) также достаточно для достоверного определения диагноза АПЖК. Достоверность диагноза определяется при наличии 2 «больших» критериев, 1 «большого» и 2 «малых» или 4 «малых» критериев дисплазии ПЖ [40]. Однако в нашем случае нет важных «больших» ЭКГ-критериев дисплазии ПЖ[39] в виде характерных изменений реполяризации миокарда в правых грудных отведениях (эпсилон-волна или инверсия зубца Т в отведениях V1—V3 при отсутствии полной БПНПГ), а морфологический паттерн выявленной неустойчивой ЖТ не соответствует форме блокады ЛНПГ (в том числе передневерхнего разветвления).
Так какой же фенотип КМП у нашей пациентки? Это бивентрикулярный подтип АПЖК со стадией болезни, имитирующей ДКМП [41, 42], или ДКМП с правожелудочковой дилатацией, дисфункцией и гипертрабекулярностью [43]. В представленном случае на этой стадии болезни довольно сложно выделить доминирующий фенотип КМП.
При ДКМП чаще поражается ЛЖ или оба желудочка с развитием дилатации всех полостей сердца и кардиомегалии [44]. Для дисплазии ПЖ характерно поражение преимущественно ПЖ, при этом кардиомегалия и легочная гипертензия наблюдаются редко [39, 40].
Основными клиническими проявлениями дисплазии ПЖ служат нарушения ритма сердца (правожелудочковая экстрасистолия, ЖТ из выходного отдела ПЖ, фибрилляция желудочков), манифестирующие признаки ламиновой ДКМП — дефекты проводящей системы, брадиаритмии, пароксизмы ЖТ и СН. Необходимо еще учитывать особенности некомпактного строения миокарда ЛЖ (в нашем случае наблюдалась бивентрикулярная гипертрабекулярность доминированием трабекулярности ПЖ, критерий отношения некомпактной и компактной частей миокарда более 2/1, но не в классической форме, когда доминирует некомпактность строения ЛЖ с незначительным вовлечением ПЖ, а наоборот). При КМП некомпактного ЛЖ часто встречаются и сопутствующие скелетные миопатии (Эмери—Дрейфуса, поясно-конечностная и др.). Но в нашем случае этот фенотип сомнителен, так как не подтверждается диагностическими критериями R. Jenni (соотношение N/C более 2, где N — некомпактный слой миокарда ЛЖ, C — компактный слой миокарда ЛЖ; отсутствие других сопутствующих аномалий сердца; наличие многочисленных, чрезмерно выступающих в полость ЛЖ трабекул с глубокими межтрабекулярными пространствами; наличие сообщающихся с полостью ЛЖ межтрабекулярных пространств, визуализируемых с помощью цветового допплер-кардиографического метода) [45].
Вторая причина — это «парадигма» строгой фенотипическо-генотипической корреляции КМП. У пациентки выявлен клинический симптомокомплекс ламиновой ДКМП: скелетно-мышечная гипотрофия с повышением уровня сывороточной КФК, клинически проявляющиеся брадиаритмии с дефектами проводимости в виде АВ-блокады (сначала проксимальная, затем дистальная) и дисфункции синусного узла, появление наджелудочковой (ФП) и желудочковой тахиаритмии. Патогномоничные клинические признаки позволили предположить диагноз ДКМП с ламинопатией, а молекулярно-генетическое исследование с выявлением миссенс-мутации в 3-м экзоне ламинового гена — верифицировать эту патологию. Но так ли все просто и однозначно? С учетом интерпретации некоторых данных МРИ и ЭхоКГ, а также результатов морфометрии образцов эксплантированного сердца вероятность ДКМП в качестве единственного фенотипа снижается. Тем более что некоторые авторы полагают, что ЛЖ сердца вовлекается в патологический процесс более чем в половине случаев заболеваний АПЖК [46—48], реже встречается преимущественное поражение ЛЖ [48—51], связанное с дефектом гена десмоплакина [51]. Известно, что и жировая инфильтрация ПЖ не считается достаточным морфологическим признаком АПЖК, так как небольшие отложения жира в эпикарде и миокарде переднебоковой и апикальной областей ПЖ, увеличивающиеся с возрастом и по мере возрастания массы тела, наблюдают у здоровых лиц [52]. В литературе последних лет встречаются множественные сообщения о клинических формах КМП со смешанными и перекрывающимися фенотипами. Авторы детально представляют клинические наблюдения с сочетающимися морфофункциональными фенотипами ГКМП и АПЖК с некомпактным строением ЛЖ, с трансформацией гипертрофического и некомпактного фенотипов в ДКМП, а также клинические ситуации с динамическими фенотипами АПЖК, некомпактной КМП и ДКМП, вызванными различными формами миокардитов [43, 53—55]. Многие исследователи полагают, что АПЖК часто сочетается как с другими генетическими КМП, так и с инфекционно-иммунным миокардитом (почти 50% больных) [53, 54, 56].
Эпидемиологические и молекулярно-генетические исследования последнего десятилетия произвели революционный переворот в понимании патофизиологии КМП и в диагностических геномных возможностях. Уже известно, что клиническая и генетическая структура ДКМП крайне сложна и гетерогенна, существуют значительные перекресты между фенотипами и генотипами. На рис. 6 графическое изображение демонстрирует генетическое разнообразие и степень «перекреста» генотипов КМП, известные к настоящему времени [5]. По данным литературы, у 5—15% пациентов с ДКМП выявляются генетические десмосомальные аномалии [57], у 6—9% — ламиновые аномалии [24]. Мутации генов, кодирующих десмосомальные белки, считаются одной из основных причин наследственных (аутосомно-доминантных) АПЖК [5, 6, 52]. Патогномоничному АПЖК фиброзно-жировому замещению кардиомиоцитов ПЖ предшествуют изменения в десмосомах и ассоциированные с ними нарушения соединений между клетками [51, 52, 57]. В 2012 г. появились первые сообщения (G. Quarta) о ламиновых мутациях, связанных с фенотипом АПЖК, и в последние годы нередко регистрируются случаи ламиновых мутаций (в том числе de novo), приводящих к развитию АПЖК и КМП некомпактного миокарда [41, 43], но ламиновые генные аномалии встречаются значительно чаще в случаях ДКМП [24].
Возможно, клиническое сходство фенотипов АПЖК и ДКМП (подтип АПЖК с бивентрикулярным поражением имитирует фенотип ДКМП, а у пациентов с ДКМП довольно часто встречаются правожелудочковая дилатация и дисфункция с гипертрабекулярностью ПЖ) может быть обусловлено их общей генетической патологией.
В 2013 г. ученые из Хельсинки и Торонто сообщили о трех новых мутациях ламина, ассоциированных с бинодальной дисфункцией и фенотипом ДКМП, по некоторым критериям сходным с АПЖК (преимущественное поражение правых отделов сердца); в том числе миссенс-мутация LMNA [р.Phe237Ser], выявленная у 5 членов семьи c наследственной формой ДКМП, по клиническим и морфофункциональным признакам оказалась похожей на представленный нами клинический случай [43].
Ситуации со смешанными и перекрестными фенотипами КМП являются сложными и неоднозначными для классификационной диагностики, так как до настоящего времени в нашей стране (как и в других странах СНГ) используется классификация КМП, принятая ВОЗ в 1995 г. Классификации Американской ассоциации кардиологов (2006) и Европейского общества кардиологов (2008) в таких клинических ситуациях также не соответствуют потребностям специалистов, так как непреодолимым ограничением всех перечисленных классификаций КМП является «перекрест» различных категорий (этиологическая причина, структурная анатомия, морфофункциональный фенотип, тип наследования, генетический дефект, функциональный статус), в которые сгруппированы эти заболевания [58, 59].
В таких случаях все сложности нивелирует новая система классификации КМП «MOGE(S)», впервые опубликованная в официальном журнале Всемирной федерации сердца (ВФС) в декабре 2013 г. Эта система отличается от всех предыдущих классификаций своей универсальностью, легкостью модификации, динамичностью, глубиной детализации и емкостью. Новая классификация включает все формы КМП, а также категорию бессимптомных носителей, ранних форм и перекрывающихся фенотипов [60]. Сами ученые признались, что на создание новой классификации их вдохновила TNM — общепринятая клиническая классификация злокачественных опухолей. Появление новой системы классификации, предложенной ВФС, свидетельствует о накоплении большого объема новой информации об этиологии КМП и глубоком изучении патофизиологических и генетических механизмов этой группы некоронарогенных заболеваний. Разработчики классификации (кардиологи, генетики, патологоанатомы и специалисты по лучевой диагностике из Америки, Италии, Канады, Индии и Австралии) предложили обновить определение КМП и рассматривать под ними «нарушения, характеризующиеся морфологическими и функциональными аномалиями миокарда на фоне отсутствия какой-либо другой болезни, способной самой по себе стать причиной развития наблюдаемого фенотипа» [60].
Система классификации получила название «MOGE (S)» по первым буквам 5 ключевых категорий: М — Morpho-functional (морфофункциональные признаки или внешние клинические проявления, так называемый клинический фенотип; О — Organ/system involvement (вовлеченные органы/системы); G — Genetic (тип наследования и доля наследственного компонента); E — Etiological Annotation (этиология — вирусная, аутоиммунная, генетическая и др.); S — Stage (стадия СН).
Основные параметры кодирования в MOGE(S) системе классификации МФС (2013 г.) представлены в таблице.
Сочетание букв и цифр позволяет создать для каждого пациента индивидуальный код заболевания, позволяющий любому участнику медицинского сообщества получить максимум информации о больном. Новая система представлена также в виде интерактивного инструмента в виде веб-приложения для врачей, которое размещено на сайте: http://moges.biomeris.com. При использовании этой системы классификации построение клинического диагноза в представленном клиническом случае становится логичным, точным, детальным и кратким, а буквенный индивидуальный код заболевания нашей пациентки будет выглядеть следующим образом: М D+A[CHB+slowAF+nsVT]O Н+М[↑sCPK] GN E G-DN-LMNA[3exon:р. Arg190Pro] SD-IV, где клинический морфофунк-циональный фенотип — М D+A[CHB+slowAF+nsVT] — представляет смешанный (перекрывающийся) вариант КМП (D — дилатационный фенотип+A — АПЖК/дисплазия), slow AF — брадисистолическая ФП с CHB (complete heart block — полная поперечная блокада сердца, симптом Фредерика); nsVT — неустойчивые пароксизмы ЖТ; O Н+М [↑sCPK] — пораженные органы: доминирующая патология сердца — H и вовлечение скелетных мышц — M с повышенным уровнем сывороточной КФК— ↑sCPK; GN — форма заболевания несемейная (ненаследственная) — N; E G-DN-LMNA[3 exon: р.Arg190Pro] этиология — генетическая, выявлен генетический дефект ядерного гена ламина в виде миссенс-мутации в 3-м экзоне (мутация de novo р.Arg190Pro); SC-III СН соответствует III стадии и IV ФК по классификации NYHA; эволюция болезни пациентки представлена на рис. 7 в виде схематической диаграммы по классификации системы MOGE.
Что же все-таки нового в данной системе классификации? Основными ее принципиальными отличиями от предшествующих перечисленных классификаций являются: а) принципиально новое определение КМП; б) впервые примененное в официальной классификации КМП буквенное кодирование одновременно 5 ключевых «перекрестных» категорий (этиология, клинико-морфологический фенотип, выявленная генетическая аномалия, механизм наследования; изолированность или системность поражения органов, стадия СН); в) возможность замены буквенного символа при изменении фенотипа КМП в течение болезни и дополнения кода символом после целенаправленного молекулярно-генетического, вирусологического, иммуногистохимического (и т.д.) обследований; г) по предложенной классификации можно достаточно кратко и полно описать сложные гено- и фенотипические проявления заболевания: смешанные и перекрывающиеся фенотипы (сочетание гипертрофического и дилатационного фенотипов — МН+D, сочетание в виде АПЖК и КМП с некомпактным строением миокарда ЛЖ — МА+NC), отличительные клинические признаки (такие, как АВ-блокада: M D [AVВ], синдром WPW: M Н [WPW], повышение уровня КФК: M D [СРК] или наличие эпсилон-волны: M A [ə], семейные бессимптомные носители: М0 и ранние формы КМП: M E [Н]); д) в системе предусмотрена возможность дополнительного введения символов, цифр и букв в индивидуальный код болезни пациента по мере появления новых данных.
С этих позиций в представленном нами случае вполне оправдано было бы проведение детального геномного анализа нашей пациентки (набор генов, ответственных за КМП), так как смешанные фенотипические формы КМП нередко имеют несколько генных мутаций, а иногда одна мутация гена связана с более чем 1 фенотипом [61—63]. И в качестве дополнительных потенциальных генов-кандидатов в нашем случае можно рассматривать все десмосомальные гены, дистрофин, тайтин и ген, кодирующий альфа-субъединицу натриевого канала (SCN5A).
В настоящее время нам представляется индивидуальный код болезни пациентки в виде МD+A[CHB+slowAF+nsVT]Oн+м[↑sCPK] GN EG-DN-LMNA[3exon:р.Arg190Pro], но вполне вероятна возможность замены этих буквенных символов после расширения спектра генетического анализа. Мы с интересом ждем новых находок в продолжающихся исследованиях.


