
В 1767 г. F. Fontana описал явление рефрактерности в сердечной мышце [1]. В 1926 г. T. Lewis и А. Drury предположили, что увеличение эффективного рефрактерного периода (ЭРП) является существенным антиаритмическим механизмом [2].
В настоящее время существует более сотни антиаритмических препаратов, которые относятся к 4 классам по классификации Vaughan—Williams [3]. Она основана на описательной картине влияния антиаритмических препаратов на трансмембранный потенциал действия (ПД). Классификация является несовершенной [4], но используется в настоящее время. Препараты, увеличивающие длительность потенциала действия (ДПД), и, соответственно, ЭРП, относятся к III классу.
Препараты III класса (дофетилид, соталол, самый распространенный в России — амиодарон) широко используются [5]. Однако они оказывают негативные эффекты и в некоторых случаях могут вызывать фибрилляцию желудочков (ФЖ) и увеличивать смертность [6]. Вызываемое ими увеличение ДПД приводит к развитию ранних постдеполяризаций (РПД) в волокнах ткани желудочков. РПД развиваются на фоне задержки реполяризации ПД (которая проявляется как удлинение интервала QT на электрокардиограмме — ЭКГ). РПД служат триггером, инициализирующим феномен re-entry, полиморфную желудочковую тахикардию (ЖТ) по типу torsade de pointes (TdP), переходящую в ФЖ. TdP впервые описал F. Dessetrenne в 1966 г. [7] как форму тахиаритмии, при которой ЭКГ приобретает характерную веретенообразную форму. TdP — основной побочный «проаритмический» эффект препаратов III класса.
Механизмы как антиаритмической, так и проаритмической активности препаратов III класса обусловлены их взаимодействиями с ионными каналами мембран кардиомиоцитов, и до конца не изучены. Этим механизмам и эффектам посвящен данный обзор.
Реполяризация в кардиомиоцитах
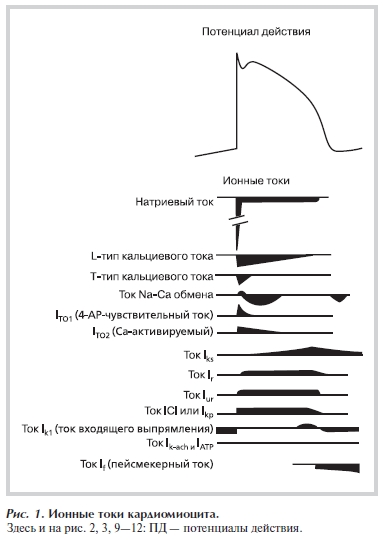
Реполяризация ПД в кардиомиоцитах определяется выходящими калиевыми токами — IKur (сверхбыс-
трый выходящий ток задержанного выпрямления), ITO1,2 (транзиторный выходящий ток), IKr и IKs (быстрый и медленный компоненты токов задержанного выпрямления), IK1 (ток аномального выпрямления) и некоторыми другими. Токи задержанного выпрямления протекают через потенциалчувствительные калиевые каналы. В настоящее время существует много литературы, посвященной классификации, описанию кинетики, молекулярной структуры, регуляции, видовому и тканевому распределению вышеуказанных токов и каналов [8, 9]. Приведем лишь схему, отражающую временное соотношение токов (деполяризующих и реполяризующих), протекающих во время ПД в «усредненном» кардиомиоците (рис. 1).
Рисунок 1. Ионные токи кардиомиоцита.
Гены калиевых каналов, переносящих ток IKr принадлежат к так называемой группе генов hERG (соответственно, каналы hERG) [10]. Утверждается, что у человека реполяризация ПД в основном определяется током IKr [11]. В настоящее время описано множество мутаций в генах hERG (более 200, см. http://www.pc4.fsm.it.81/cardmoc/), приводящих либо к ослаблению, либо к усилению IKr и других калиевых токов. Мутации в генах именно этих каналов являются причиной наследственных синдромов, проявляющихся в удлинении (LQTx) либо уменьшении (SQTx) интервала QT на ЭКГ. Важно, что увеличение интервала QT и его уменьшение тесно связаны с различными нарушениями ритма сердца
и ФЖ [12]. Агонисты и блокаторы калиевых каналов задержанного выпрямления изменяют длительность ПД и интервала QT. Препараты III класса являются блокаторами указанных каналов.
На рис. 2 схематически показаны интервалы QT и ПД желудочкового кардиомиоцита в норме, при синдромах LQTx и SQTx, при действии блокаторов и активаторов каналов задержанного выпрямления.
Рисунок 2. Калиевые токи задержанного выпрямления (IKr, IKs) определяют длительность ПД в желудочке и интервала QT на ЭКГ.
Усиление калиевых токов задержанного выпрямления (например, при усилении экспрессии генов, кодирующих канальные белки) приводит к снижению ДПД и ЭРП, уменьшению длины волны возбуждения и, соответственно, к увеличению вероятности образования контура циркуляции волны возбуждения, мерцанию и трепетанию предсердий, ФЖ [13, 14].
В большом количестве работ показано, что при различных патологических состояниях, например сердечной недостаточности, в постинфарктный период, при гипокалиемии, при гипертрофии миокарда в желудочке значительно снижаются реполяризующие токи IKr, IKs, IK1. Все эти состояния сопровождаются повышенной вероятностью нарушений ритма. Введение при таких состояниях блокаторов IKs, IKr увеличивает вероятность развития постдеполяризаций, re-entry, TdP, ФЖ [15, 16].
Таким образом, как усиление, так и подавление реполяризующих токов приводит к опасным последствиям. Увеличение ДПД и замедление реполяризации в результате применения препаратов III класса не является однозначным явлением. В следующих разделах обзора обсуждаются условия, при которых могут проявляться их анти- и проаритмические эффекты.
«Морфология» потенциала действия и аритмии
Увеличение ДПД может происходить в результате нескольких процессов.
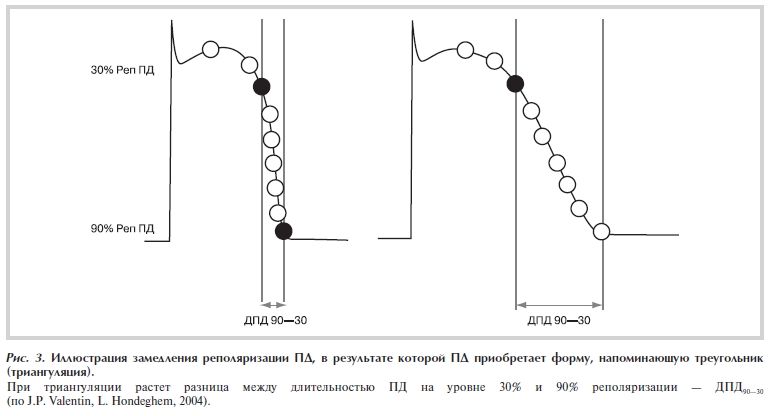
А. ДПД может быть увеличена в результате задержки реполяризации — удлиняется фаза плато ПД, а время реполяризации от уровня плато до уровня потенциала покоя (ПП) не изменяется.
Рисунок 3. Иллюстрация замедления реполяризации ПД, в результате которой ПД приобретает форму, напоминающую треугольник (триангуляция).
Б. ДПД может увеличиваться за счет замедления реполяризации (длительность фазы плато не меняется, но происходит удлинение 3-й фазы ПД — время реполяризации от уровня плато до уровня ПП увеличивается). Замедление реполяризации приводит к тому, что ПД приобретает форму треугольника. Для обозначения такого преобразования ПД L. Hondeghem и соавт. предложили термин «триангуляция» [17].
В свою очередь триангуляция ПД может развиваться на фоне 1) суммарного увеличения; 2) снижения или 3) без изменения длительности ПД. При исследовании большой группы блокаторов токов задержанного выпрямления было показано, что 60% из них увеличивают ДПД, а 40% снижают ДПД в желудочках и волокнах Пуркинье [18]. Триангуляция может быть обусловлена 1) подавлением реполяризующих токов IKr, IKs, Ik1; 2) усилением тока Na—Ca-обменника — INa—Ca; 3) подавлением деполяризующих токов IСa,L.
Степень триангуляции можно определить, рассчитав разницу ДПД на уровне 90% и 30% реполяризации (ДПД90—30) (рис. 3). По данным многочисленных исследований, триангуляция независимо от ее механизмов и причин, свидетельствуют об увеличении вероятности развития аритмий [19—21]. На ЭКГ триангуляция проявляется как уплощение и расширение волны Т и может сопровождаться увеличением интервала QT. Существует несколько причин, по которым триангуляция является проаритмическим фактором.
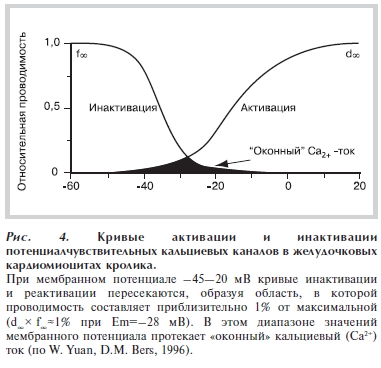
Рисунок 4. Кривые активации и инактивации потенциалчувствительных кальциевых каналов в желудочковых кардиомиоцитах кролика.
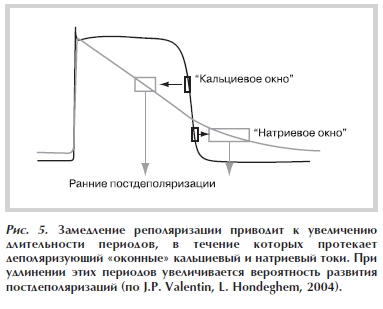
А. Снижение скорости реполяризации ПД приводит к увеличению длительности «кальциевого окна» — времени, когда мембранный потенциал находится в диапазоне от –45 до –20 мВ. В этот период кальциевые каналы, переносящие деполяризующий IСa,L, еще не полностью инактивированы (или уже частично реактивированы), и их проводимость составляет около 1% от максимальной — протекает так называемый оконный кальциевый ток [22] (рис. 4). Задержка реполяризации способствует реактивации кальциевых каналов и увеличивает время, в течение которого протекает входящий деполяризующий кальциевый ток (ICa,L). Предполагается, что за счет этого тока может возникнуть РПД (рис. 5).
Рисунок 5. Замедление реполяризации приводит к увеличению длительности периодов, в течение которых протекает деполяризующий «оконные» кальциевый и натриевый токи. При удлинении этих периодов увеличивается вероятность развития постдеполяризаций (по J.P. Valentin, L. Hondeghem, 2004).
Рисунок 6. Состояния ионных потенциалчувствительных каналов и переходы между ними.
При действии β-адреномиметиков, гипертрофии миокарда увеличивается «остаточный» кальциевый ток, т.е. проводимость, наблюдающаяся после окончания пикового компонента тока. β-Адреномиметики изменяют кинетику кальциевых каналов: ускоряют их реактивацию [23]
после инактивации, соответствующей концу фазы плато ПД. На фоне замедления реполяризации эти эффекты будут способствовать возникновению РПД [24]. Б. Снижение скорости реполяризации ПД приводит к увеличению длительности периода времени, в течение которого Na-потенциалчувствительные каналы находятся в частично реактивированном состоянии,
а мембранный потенциал еще не достиг уровня ПП — так называемое натриевое окно (соответствующее части периода относительной рефрактерности). «Натриевое окно» лежит в области более отрицательных значений мембранного потенциала, чем «кальциевое окно»
(см. рис. 5). Увеличение длительности «натриевого окна» способствует возникновению РПД по тем же причинам, что и увеличение «кальциевого окна» [25, 26].
В. Одной из причин триангуляции является усиление INa—Ca [27]. Во время 3-й фазы реполяризации Na—Ca-обменник работает в прямом режиме, т.е. выводит один ион Са2+ из клетки и вводит в обмен 3 иона Na+, тем самым генерируя входящий деполяризующий ток. Этот ток приводит к дополнительному замедлению реполяризации. Предполагается, что в ряде случаев деполяризующая компонента INa—Ca прямо приводит к развитию ранних и задержанных постдеполяризаций [27]. С увеличением ДПД (например, при действии препаратов III класса) время, в течение которого INa—Ca оказывает деполяризующее воздействие, увеличивается, и, соответственно, увеличивается вероятность постдеполяризаций.
Увеличение INa—Ca может быть обусловлено перегрузкой цитоплазмы кардиомиоцита Са2+, например, при действии β-адреномиметиков. Усиление INa—Ca наблюдается при сердечной недостаточности, инфаркте миокарда и других патологических состояниях [15].
Г. Увеличение ДПД приводит к задержке возвращения мембранного потенциала к уровню ПП
(уменьшению длительности диастолического интервала) и наступления момента, когда потенциалзависимые каналы будут полностью реактивированы. В результате к началу деполяризации следующего ПД не все натриевые каналы способны активироваться. Скорость нарастания фронта следующего ПД, скорость проведения и длина волны возбуждения в следующем цикле будут снижены. Эти эффекты облегчают образование контуров re-entry [28].
Д. К триангуляции ПД может приводить подавление выходящих калиевых токов. Подавление реполяризующих токов облегчает возникновение РПД, TdP, re-entry [28, 29].
Е. В результате гетерогенности миокарда величина Ikr, Iks имеет разброс в различных областях желудочка (как по поверхности, так и в трансмуральном направлении). Большинство блокаторов токов задержанного выпрямления не являются строго селективными и оказывают влияние как на IKr, так и на IKs. Как правило, триангуляция сопровождается значительным увеличением разброса длительностей ПД [28, 29]. Согласно некоторым теориям, разброс ДПД является основой для возникновения нарушений ритма [30, 31].
Как уже отмечалось, связь нарушений ритма (РПД, TdP и д.р.) и триангуляции подтверждена во многих
исследованиях [20, 32]. Существенно, что большинство препаратов III класса увеличивают ДПД, замед-
ляя реполяризацию, т.е. вызывают «триангуляцию» ПД. На фоне такого влияния на ПД начинают оказывать воздействие практически все вышеперечисленные проаритмические факторы (А—Е). Следует отметить, что факторы, вызывающие увеличение скорости реполяризации и снижающие ДПД90—30 (т.е. эффект, противоположный «триангуляции»), снижают и вероятность нарушений ритма [32, 33].
Особенности кинетики IKr, IKs Согласно общепринятым представлениям потенциалчувствительные ионные каналы могут находиться в открытом, закрытом и инактивированном состояниях, между которыми возможны определенные переходы (рис. 6).
Особенности кинетики каналов, переносящих IKr и IKs, таковы, что во время деполяризации (фаза 0), во время начальной реполяризации и плато (фазы 1 и 2) ПД каналы переходят в инактивированное состояние за счет «быстрой потенциалзависимой инактивации» [34]. В ходе реполяризации ПД, когда мембранный потенциал становится чуть отрицательнее уровня плато ПД (≈–30 мВ), «быстрая инактивация» сменяется «медленной потенциалзависимой реактивацией», т.е. только с окончанием
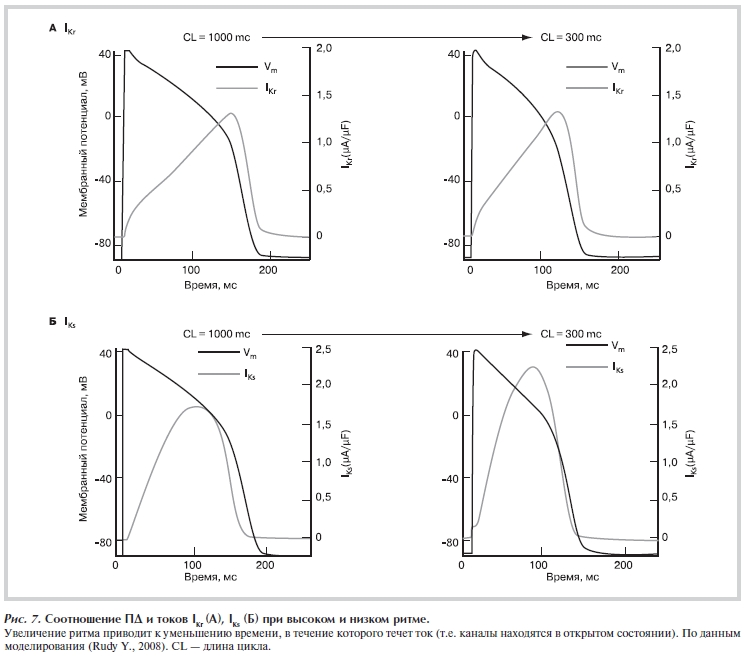
фазы плато ПД канал переходит в открытое состояние. К концу фазы плато ПД ток IKr достигает максимума, а потом начинает снижаться. Каналы за счет «медленной деактивации» переходят в закрытое состояние. Таким образом, токи IКr и IKs имеют выраженный пик, соответствующий концу фазы плато ПД (рис. 7, а).
Рисунок 7. Соотношение ПД и токов IKr (а), IKs (б) при высоком и низком ритме. Увеличение ритма приводит к уменьшению времени, в течение которого течет ток (т.е. каналы находятся в открытом состоянии). По данным моделирования (Rudy Y., 2008). CL — длина цикла.
В момент окончания фазы плато ПД значение мембранного потенциала определяется тонким балансом между деполяризующими (IСaL, «хвостовым» INa, INa-Ca) и реполяризующими (в основном IKr, IKs) токами. Незначительное изменение баланса этих токов будет приводить к значительному сдвигу мембранного потенциала. Этот момент соответствует «кальциевому окну». Поэтому небольшое снижение или задержка реполяризации (например, за счет подавления IKr препаратами III класса) может способствовать возникновению РПД [34, 35].
Ток задержанного выпрямления IKs имеет схожую с IKr кинетику — его пик приходится на 3-ю фазу ПД
(рис. 7, б). В отличие от человека, IKs и IKr у кролика, собаки, морской свинки вносят приблизительно одинаковый вклад в реполяризацию ПД [10, 34]. Видовые различия затрудняют исследования эффектов блокаторов токов (и препаратов III класса в частности).
Зависимость эффекта препарата от ритма
Возникает следующий вопрос: одинаковы ли эффекты блокаторов токов задержанного выпрямления при различном ритме, и как их эффекты зависят от ритма?
В 70-х годах прошлого столетия Кацунгом и Хондегхемом была предложена «теория модулированного сродства рецептора» [36—38], которая описывала взаимодействие блокатора с ионным каналом. Согласно данной теории, определенный блокатор (фармакологический препарат) может связываться с каналом, находящимся в определенном состоянии: открытом, закрытом либо инактивированном. В настоящее время теория «модулированного рецептора» наглядно подтверждена экспериментальными данными [39].
При изменении ритма сердца время пребывания канала в том или ином состоянии изменяется. Соответственно меняется доступность канала для воздействия вещества, и величина эффекта блокатора будет зависеть от ритма сердца [29]. Возможны 2 варианта событий: при увеличении ритма эффект увеличивается (прямая зависимость эффекта от ритма), или, наоборот — снижается (обратная
зависимость эффекта от ритма).
Такие препараты III класса, как соталол, дофетилид характеризуются обратной зависимостью эффекта от ритма. При низком ритме они значительно увеличивают ДПД и интервал QT, а при высоком ритме слабо изменяют эти параметры. Это значит, что они могут способствовать РПД и TdP при низком ритме, а при высоком ритме будут оказывать слабое антиаритмическое действие [40—42]. Вероятность возникновения TdP больше при низком ритме, чем при высоком.
Показано, что все препараты, при действии которых может возникать TdP, обладают обратной зависимостью эффекта от ритма. И наоборот: если некое новое соединение характеризуется обратной зависимостью эффекта от ритма, то существует вероятность, что оно будет вызывать TdP [29].
В случае hERG-каналов обратная зависимость эффекта от ритма обусловлена двумя причинами. Первое: предполагается, что большинство блокаторов связываются с каналами, находящимся в открытом состоянии. Второе: при снижении частоты сокращений время пребывания каналов в открытом состоянии увеличивается, а при увеличении частоты — снижается [34] (см. рис. 7).
Среди блокаторов hERG-каналов, (помимо препаратов III класса) встречаются вещества, относящиеся
к самым разнообразным группам — антигистаминные средства (флюоксетин), антибиотики (эритромицин), психотропные препараты (галоперидол) [43]. Такое разнообразие свидетельствет о некой особенности каналов, делающей их более «восприимчивыми», чем другие, к соединениям, имеющим разнообразную структуру. Предполагается, что hERG-каналы обладают «универсальным» сайтом связывания, и что во всех случаях механизм блокирования сходен [44, 45].
Установлено, что сайт связывания располагается внутри поры канала. Пора со стороны цитоплазмы
имеет расширение, внутрь которого обращены 8 остатков аминокислот, имеющих ароматическую структуру. Эти остатки располагаются внутри расширения в виде 2 концентрических кругов (4 остатка в каждом «круге», один круг над другим). Такая структура способна легко образовывать связи с разнообразными органическими молекулами. Блокаторы связываются только в случае, когда канал находится в открытом состоянии. При закрытии или инактивации канала конфирмационные
изменения приводят к диссоциации блокатора [45, 46].
Как указано выше, при увеличении ритма время пребывания hERG-каналов в открытом состоянии уменьшается. Такое поведение каналов является одним из механизмов частотной адаптации ПД (rate adaptation, APD adaptation) — явления, за счет которого происходит укорочение ПД (за счет 2-й и 3-й фазы) при увеличении ритма [47]. Поведение hERG-каналов при изменении ритма описывается достаточно сложными биофизическими моделями. На качественном уровне его можно описать следующим образом. Каналы могут находиться в «глубоких» и «неглубоких» закрытых состояниях. Переходы между состояниями каналов являются время- и потенциалзависимыми процессами, и обусловлены свойствами сенсора потенциала (рис. 8, см. цветную вклейку). Для открытия канал должен пройти через последовательность закрытых состояний — от наиболее «глубокого» закрытого к открытому. При высоком ритме каналы накапливаются в «неглубоких» закрытых состояниях, из которых во время последующего ПД с большей вероятностью и более быстро переходят в открытое состояние. Соответственно при высоком ритме амплитуда реполяризующих калиевых токов IKr, IKs имеет большую амплитуду. За счет этого реполяризация ускоряется, длительность ПД снижается и каналы быстрее возвращаются в закрытое состояние [34, 35, 47—49], т.е. суммарное время их пребывания в открытом состоянии уменьшается (см. рис. 7, б).
Помимо «триангуляции» ПД и ритмозависимости, существует еще несколько причин, которые могут способствовать возникновению нарушений ритма при действии фармакологических препаратов.
Осцилляции ДПД
ДПД может меняется от цикла к циклу в одной точке тогда, когда ритм остается постоянным (при постоянной длительности цикла), т.е. могут наблюдаться осцилляции ДПД (ритмическая вариабельность ДПД). Осцилляции ДПД способствуют развитию аритмий. Предполагается, что существует критическая амплитуда осцилляций, превышение которой связано с развитием фибрилляции. Чем больше величина осцилляций ДПД, тем больше вероятность возникновения
циркуляции волны возбуждения [50].
В нормальных условиях осцилляции ДПД появляются при достижении определенного уровня ритма.
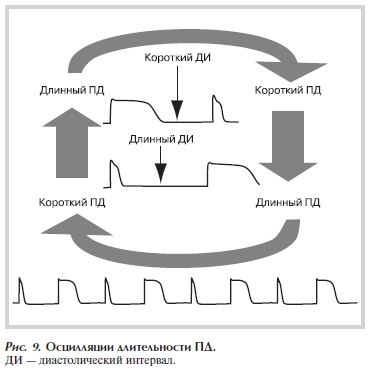
Очередная волна возбуждения приходит в некую точку в тот момент, когда ткань находится в состоянии относительной рефрактерности после предыдущего возбуждения (например, за счет того, что ионные каналы не полностью реактивированы, цитоплазматическая концентрация Са2+ не восстановлена до уровня, соответствующего покою). При осцилляции ДПД за «длинным» ПД следует короткий диастолический интервал (ДИ). После «короткого» ДИ следует «короткий» ПД и «длинный» ДИ т.д. (рис. 9).
Рисунок 9. Осцилляции длительности ПД.
Механизмы возникновения осцилляций ДПД сложны, исследованы не полностью и обусловлены взаимодействием множества факторов. Например, величина ритма, при которой будут возникать осцилляции (точка осцилляции — break point), разница между длинным и коротким ПД (амплитуда осцилляций) зависят от множества факторов: скорости изменения ритма, работы Na—Ca-обменника и саркоплазматической кальциевой АТФазы, уровня фосфорилирования кальциевых каналов, уровня
цитоплазматического Са2+, кальцийзависимого ингибирования кальциевых каналов [51].
Фармакологические препараты, особенно препараты III класса, могут вызывать или усиливать осцилляции ДПД, сдвигать точку осцилляции, так как Ikr и Iks определяют скорость реполяризации ПД, длительность ПД и длительность рефрактерного периода [17, 18, 28]. При исследовании «безопасности» препаратов могут быть оценены такие параметры, как сдвиг «точки осцилляции» ДПД, влияние на величину осцилляций ДПД, зависимость «точки осцилляции» от скорости изменения ритма и др.
Показано, что препараты, характеризующиеся обратной зависимостью эффекта от ритма, с большой вероятностью будут вызывать осцилляции ДПД. Экспериментально показано, что если препарат, начиная с некоторой концентрации, увеличивает длительность интервала QT, вызывает РПД и TdP, то осцилляции ДПД наблюдаются при использовании данного препарата в 100 раз в меньшей концентрации [17]. Эту особенность можно применять при разработке и тестировании антиаритмических препаратов. Фармакологические препараты могут вызывать осцилляции ДПД либо только при низком ритме, либо только при высоком. Например, теродилин вызывает вариабельность ДПД только при низком ритме, другой препарат — цизаприд — только при высоком [52].
Пространственный разброс ДПД
Длительность ПД при постоянном ритме не одинакова в различных областях миокарда, т.е. наблюдается пространственный разброс (или дисперсия) ДПД. Длительность ПД варьирует в апикально-базальном, транссептальном направлении и в разных слоях миокарда — трансмуральная дисперсия ДПД (transmural dispersion of repolarisation — TDR). Связи пространственной дисперсии ДПД и нарушений ритма посвящено большое количество работ (начиная с работ G.K. Moe [53]). В общем случае утверждается, что увеличение разброса ДПД коррелирует с увлечением вероятности аритмии [31]. Собственно дисперсия ДПД не является причиной ЖТ или ФЖ. Однако при наличии некоего триггера (постдеполяризаций, внеочередного стимула и т.д.) дисперсия ДПД существенно облегчает развитие TdP, re-enty и ФЖ. Причиной разброса ДПД является неоднородность миокарда по таким параметрам, как уровень экспрессии и свойства щелевых контактов, ионных каналов, обменников и АТФаз [54, 62].
Согласно преобладающей в настоящее время гипотезе, нарушению ритма (TdP и ФЖ) наибольшим образом способствует трансмуральный разброс ДПД [31, 55, 56]. Трансмуральная дисперсия ДПД обусловлена различиями по ДПД в эпикардиальных, интамуральных и эндокардиальных кардиомиоцитах. Большую ДПД имеют кардиомиоциты среднего слоя миокарда (так называемые М-клетки). Эта их особенность обусловлена меньшей амплитудой тока IKs; большей амплитудой позднего компонента тока INa и тока INa—Ca по сравнению с эпи- и эндокардиальными клетками
[57]. Соотношение токов IKr/IKs в М-клетках отличается от такового в поверхностных слоях миокарда [56].
ПД М-клеток в ответ на снижение ритма или при действии веществ, увеличивающих ДПД [58—60], растет в большей степени, чем ПД эпи- и эндокардиальных клеток [61]. Таким образом, препараты III класса могут способствовать росту трансмуральной дисперсии ДПД (например, соталол) [56].
Некоторые препараты могут уменьшать дисперсию ДПД. Предполагается, что таким препаратом является амиодарон [63] (из некардиотропных — пентобарбитал [64]). Однако в ряде работ показано, что в некоторых условиях амиодарон увеличивает трансмуральную дисперсию ДПД и может вызывать проаритмические эффекты. В субмикромолярных концентрациях этот препарат усиливает ICa,L, увеличивает длительность «кальциевого окна», амплитуду «хвостового» компонента тока INa (хотя подавляет «пиковый» компонент INa), что ведет к росту разброса ДПД в миокарде [61, 63]. Результирующий эффект зависит от концентрации
вещества и состояния миокарда.
Как и в случае с амиодароном, большинство веществ не являются строго селективными и влияют одновременно на несколько мишеней в сердце. Поэтому прогнозировать степень влияния на дисперсию ДПД практически невозможно.
На основе работ, проведенных в нашей лаборатории, можно утверждать, что в желудочках существует значительная апикально-базальная дисперсия ДПД. Этот тип дисперсии наряду с трансмуральной может играть значительную роль в нарушении ритма [65].
Помимо дисперсии ДПД, в сердце имеется дисперсия скорости проведения возбуждения (Vпров), длины волны возбуждения (λ). В среднем слое миокарда снижена плотность щелевых контактов (Cx43), изменена ориентация клеток относительно поверхностных слоев [62], что увеличивает сопротивление ткани и вносит вклад в рост трансмуральной дисперсии Vпров. Наблюдается пространственная дисперсия степени триангуляции, частотной «эффективности», «нестабильности ДПД», т.е. разброс имеет практически любой электрофизиологический параметр, который можно оценить.
Взаимодействие электрофизиологических параметров
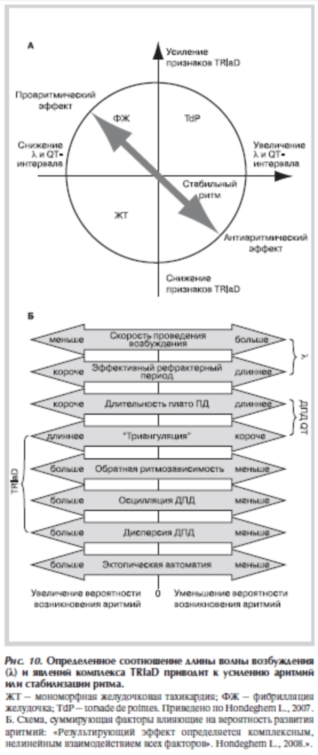
Триангуляция ПД, обратная зависимость эффекта от ритма, осцилляции ДПД, пространственная дисперсия ДПД — взаимосвязанные явления, имеют общие механизмы и чаще всего возникают совместно. Поэтому их объединяют в группу (или комплекс), которая в англоязычной литературе условно обозначается TRIaD (Triangulation, Reverse-use-dependance, Instability, APD dispersion) [17, 28, 29, 33].
Согласно классическим представлениям, увеличение ДПД и рефрактерного периода являются антиаритмическими эффектами. В то же время связанное с увеличением ДПД удлинение интервала QT коррелирует с TdP и ФЖ. Возникает противоречие, которое разрешается только с применением представления о TRIaD. Если увеличение ДПД сопровождается явлениями комплекса TRIaD, то эффект некого препарата или воздействия будет проаритмическим. Если ДПД увеличивается, а проявления комплекса TRIaD снижаются, то эффект может быть антиаритмическим.
Помимо TRIaD, необходимо учитывать изменения длины волны возбуждения λ [произведение скоро-
сти проведения возбуждения (Vпров) и ЭРП]. λ зависит от скорости нарастания фронта ПД и ДПД и, безусловно, связана с TRIaD. Однако λ может как уменьшаться, так и увеличиваться при появлении комплекса TRIaD, и в этом плане является дополнительным, относительно независимым параметром. Всего возможны 4 варианта комбинации TRIaD и λ.
1. Наиболее опасна такая ситуация, когда появляются признаки комплекса TRIaD и происходит снижение λ, например, при снижении Vпров совместно с триангуляцией ПД (в результате укорочения фазы плато ПД) [66]. В этом случае значительно растет вероятность ФЖ за счет уменьшения ЭРП [67]. Следует учесть, что снижение λ не приводит к аритмиям, но облегчает образование контуров циркуляции волны возбуждения [68]. 2. В случае, если одновременно увеличивается λ и усиливаются проявления TRIaD, наиболее вероятно появление TdP. Причиной TdP в данных условиях являются
постдеполяризации и временная вариабельность ДПД, которая создает возможность для циркуляции волны возбуждения при большой λ. Большая длительность ПД и рефрактерного периода в этом случае препятствует возникновению ФЖ. Однако переход TdP в ФЖ все же возможен. Поэтому данная комбинация λ и TRIaD находится на втором месте по опасности [66].
Видимо, вероятность возникновения TdP при увеличении ДПД меньше, чем вероятность развития ФЖ, при снижении ДПД. Однако ФЖ практически всегда имеет летальный исход, соответственно, вероятность выявления «фармакологических» ФЖ гораздо ниже, чем TdP.
3. Если λ снижается, но увеличения TRIaD не происходит или проявления TRIaD уменьшаются (т.е. ДПД
снижается за счет ускорения реполяризации), тогда велика вероятность развития стабильных аритмий,
например мономорфной ЖТ.
4. Только одновременное увеличение (или отсутствие снижения) λ и уменьшение проявления TRIaD не будет приводить к возникновению аритмий (Vпров не снижается, ДПД и рефрактерный период увеличиваются, скорость реполяризации ПД увеличивается).
Выше приведены только крайние варианты соотношения λ и TRIaD. Однако соотношение данных
параметров может изменяться достаточно плавно, оно может изменяться во времени. Поэтому соотношение параметров и конечный эффект удобно представить на круговой диаграмме, которая приведена на рис. 10, а. На рис. 10, б приведена схема, суммирующая факторы, влияющие на вероятность развития аритмий.
Рисунок 10. Определенное соотношение длины волны возбуждения (λ) и явлений комплекса TRIaD приводит к усилению аритмий или стабилизации ритма.
Интервал QT как проаритмический предиктор
Электрокардиографическим проявлением эффектов препаратов III класса в желудочке является увеличение интервала QT. В 1994 г. в результате программы SWORD [69] было показано, что при удлинении интервала QT увеличивается смертность. Появление «фармакологического» TdP коррелирует с увеличением длительности интервала QT на ЭКГ [70]. С некоторого времени удлинение интервала QT стало синонимом проаритмической активности. Поэтому в ходе доклинических и клини-
ческих испытаний любой новый препарат исследуется на способность увеличивать длительность интервала QT. В США испытания проводятся согласно предписанию «Оценка потенциала к увеличению интервала QT среди некардиотропных медицинских продуктов». До 1997 г. считалось, что препарат безопасен, если увеличивает интервал QT не более чем на 30—60 мс. С 2005 г. вступили в силу новые правила, согласно которым препарат влияет на интервал QT, если увеличивает его на 5 мс, и считается «опасным», если увеличивает на 20 мс (www.fda.gov/CbER/gdlns/iche14qtc.htm).
Однако увеличение интервала QT, корригированного интервала QT (QTc) и даже увеличение разброса QT не являются надежными признаками, указывающими на рост вероятности TdP, ЖТ, ФЖ. TdP может возникать без увеличения интервала QT, и наоборот, увеличение интервала QT может не приводить к TdP. Использование параметра «длительность интервала QT» может приводить к появлению ложноположительных («нет увеличения интервала QT, следовательно, нет проаритмического эффекта»), и ложноотрицательных («есть увеличение интервала QT, следовательно увеличивается вероятность аритмий») результатов [28, 29, 33].
Градиент потенциала, являющийся результатом разного временного хода реполяризации в эндо-, интра- и эпикардиальных кардиомиоцитах, служит причиной образования волны Т на ЭКГ. Величина разброса ДПД пропорциональна длительности периода Тпик—Тконец волны T. В ряде работ, выполненных под руководством С. Antzelevich утверждается, что Тпик—Тконец является единственным надежным параметром ЭКГ («макроскопическим»), на основе которого можно судить об антиили проаритмическом действии вещества [61, 71—73].
Однако для понимания механизмов действия и возможных эффектов препарата данных о его влиянии
на интервал QT недостаточно. В ходе доклинического исследования препарата на животных можно получить данные не только о его влиянии на длительность интервала QT, но и на множество электрофизиологических параметров, связанных с TRIaD и λ.
В качестве примера можно привести теродилин и толтеродин — препараты, которые предполагалось использовать при расстройствах мочеиспускания. Оба препарата в субтерапевтических концентрациях блокируют hERG-каналы. Теродилин вызывал TdP, но не увеличивал интервал QT, в то время как толтеродин не вызывал TdP, но увеличивал интервал QT. С помощью микроэлектродной техники было показано, что теродилин не вызывает увеличения ДПД, но снижает длительность плато и замедляет реполяризацию, т.е. вызывает триангуляцию ПД. Толтеродин вызывает увеличение ДПД на уровне фазы плато, но не замедляет реполяризацию. С помощью техники фиксации потенциала было показано, что теродилин вызывал одинаковое снижение тока IKr при потенциале, равном 0 и –30 мВ (что соответствует фазе плато и началу фазы реполяризации ПД). Толтеродин снижал IKr толь-
ко при потенциале равном, 0, а при потенциале –30 мВ усиливал (т.е. способствовал удлинению фазы плато ПД, но ускорял фазу реполяризации) [29, 74]. Таким образом, указанные препараты, блокируя один и тот же ионный ток, вызывали совершенно разные эффекты (рис. 11). Различия в эффектах, видимо, обусловлены тем, что препараты взаимодействуют с калиевыми каналами, когда те находится в различных состояниях.
Рисунок 11. Различие эффектов двух блокаторов IKr — теродилина и толтеродина.
Такие препараты, как карведилол (адреноблокатор), эбастин, лоратадин (антигистаминные средства),
ранолазин (антиангинальный препарат), сальбутамол (β-адреномиметик), тамоксифен (противоопухолевый препарат) в терапевтических концентрациях удлиняют интервал QT, но не вызывают увеличения признаков TRIaD и поэтому допущены к применению [17, 28, 29, 33].
Перспективы разработки новых антиаритмических препаратов
В настоящее время вопрос о разработке новых антиаритмических препаратов остается актуальным, поскольку существующие препараты недостаточно эффективны и оказывают выраженные побочные действия. В полной мере это касается препаратов III класса, которые в основном применяются при фибрилляции предсердий (ФП) для восстановления и сохранения синусового ритма. Можно выделить два основных положения, характеризующих прогресс в данной области — безопасность и селективность.
Иллюстрацией к первому положению служат дронедарон и азимилид. Эти препараты являются производными амиодарона, но не содержат атомов йода. Соответственно эти препараты не вызывают нарушений функционирования щитовидной железы. Дронедарон и азимилид менее липофильны, чем амиодарон. Эти соединения в меньшей степени накапливаются в тканях и быстрее выводятся почками и поэтому обладают меньшей токсичностью. Тем не менее дронедарон повышает смертность при сердечной недостаточности. Как и амиодарон, его производные подавляют несколько ионных токов — INa, IСa, IKr, IKs, IK1, IKAch, IKur. Механизм антиаритмического действия дронедарона и азимилида, видимо, обусловлен сложным соотношением влияний этих соединений на каналы, переносящие указанные токи [75].
Другой путь развития заключается в повышении селективности препаратов. Для восстановления синусового ритма при ФП необходимо увеличение ДПД в предсердиях, но этого не должно происходить в желудочках. Селективные блокаторы каналов, переносящих токи, характерные только для предсердий, могли бы оказывать такое действие, являться эффективными антиаритмическими средствами, и не вызывать проаритмические эффекты в желудочках.
На эту роль не подходят блокаторы IKr, IKs (эти токи имеются и в предсердиях, и в желудочке). Для предсердий человека характерны такие реполяризующие токи, как IKur, ITO.1, IKAch (ацетилхолинзависимый ток) [15, 76]. Блокаторы каналов, переносящих эти токи, могут быть
эффективными антиаритмическими препаратами.
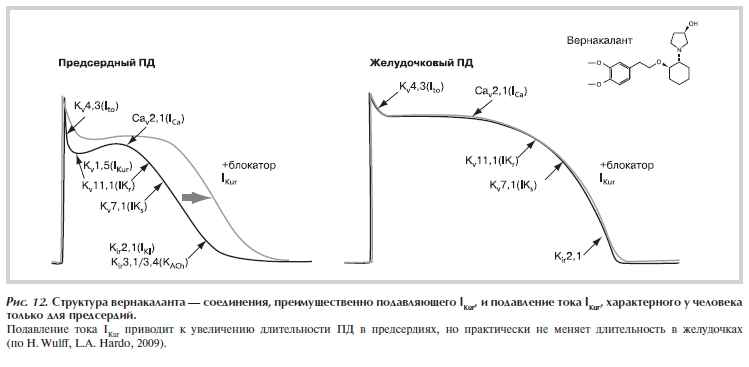
Рисунок 12. Структура вернакаланта — соединения, преимущественно подавляющего IKur, и подавление тока IKur, характерного у человека только для предсердий.
Показано, что в предсердиях существует базальный уровень IKach, т.е. данный ток наблюдается в отсутствие парасимпатического тонуса и действия холиномиметиков. При ремоделировании миокарда, вызванном хронической ФП, амплитуда базального компонента IKach возрастает. Возможно, что эта базальная составляющая тока играет ключевую роль в поддержании ФП. Вероятно, отчасти механизмы антиаритмического действия амиодарона и дронедарона обусловлены подавлением IKach. Предполагается, что селективные блокаторы предсердного IKach будут оказывать антиаритмическое действие [15, 77, 78].
Особое внимание уделяется блокаторам IKur (ток, переносимый каналом Kv1.5). Этот ток определяет раннюю реполяризацию ПД в предсердиях. Показано, что при действии соединений, преимущественно блокирующих IKur, ДПД в предсердиях может существенно увеличиваться, а в желудочках не меняться (рис. 12). Примером такого соединения является вернакалант. Показано, что при внутривенном введении вернакаланта происходит восстановление синусового ритма при ФП в 4—61% случаев (при дозе 2 мг/кг). Утверждается, что вернакалант не вызывает TdP и других проаритмических осложнений [15, 75].
Соединения, подобные вернакаланту активно разрабатываются такими фирмами, как Procter and Gamble, Merck, Sanofi-Aventis, Pfizer. Исследования затруднены тем, что у мышей, крыс кроликов, собак и у человека IKur имеет различную выраженность, распределение в сердце и чувствительность к блокаторам. Исследования, выполненные на разных животных, и в разных условиях, дают противоречивые результаты. В настоящее время запатентованы более 50 соединений, но все они
не являются «абсолютно» селективными для IKur и в той или иной степени блокируют IKr, IKs, INa, ICa. Вернакалант, преимущественно блокирует IKur, но также является блокатором INa и ITO. Большинство этих соединений не прошли стадии испытаний на животных, и только вернакалант прошел III стадию испытаний и, вероятно, будет рекомендован Управлением по контролю за качеством лекарственных препаратов и пищевых продуктов США как препарат для восстановления синусового ритма при ФП (внутривенное применение) [79].
Отечественный препарат РГ-2
В заключение отметим отечественный препарат РГ-2. В ходе доклинической фазы электрофизиологических исследований показано, что РГ-2 увеличивает ДПД в предсердных кардиомиоцитах крысы и кролика (в концентрации 1 мкМ на 26±7%). Это действие связано с подавлением калиевых токов задержанного выпрямления. Препарат увеличивает ЭРП в предсердиях собаки in situ, начиная с дозы 1 мкг/кг, а в желудочке — только при дозе 40 мкг/кг. Увеличение интервала QTc начиналось только с дозы 20 мкг/кг и, вероятно, обусловлено замедлением синусового ритма (РГ-2 вызывает увеличение интервала RR на ЭКГ). Этот эффект, видимо, связан с небольшим снижением ICa, L при действии РГ-2. По предварительным данным, РГ-2 не вызывает развития TdP в диапазоне исследованных доз.
Степень увеличения ЭРП под действием РГ-2 не зависела от исходной частоты стимуляции, т.е. препарат не обладает обратной зависимостью эффекта ритма от частоты сердечных сокращений, характерной для многих препаратов III класса и связанной с проаритмическими эффектами. РГ-2 не изменяет скорость проведения возбуждения в предсердиях. Вероятно, РГ-2 не является блокатором INa, который определяет скорость проведения возбуждения. Эти данные свидетельствуют в пользуРГ-2 по сравнению с производными амиодарона и вернакалантом. РГ-2 обладает антихолинергической активностью — ослабляет укорочение предсердных ПД, вызванное действием холиномиметиков. Подобное действие может быть особенно важно при зависимых от блуждающего нерва суправентрикулярных тахиаритмиях. РГ-2 восстанавливает и поддерживает синусовый ритм после
прекращения фибрилляции предсердий, вызванной стимуляцией блуждающих нервов у собак in situ. При использовании препарата в дозах 20 и 40 мкг/кг прекращение ФП происходило в 100% случаев. Предотвращение фибрилляции наблюдалось в 80% случаев.
Совокупность электрофизиологических данных, которые опубликованы более чем в 25 работах [80], свидетельствует о том, что РГ-2 может быть отнесен к препаратам III класса. Механизм антиаритмического действия РГ-2, вероятно, обусловлен преимущественным его влиянием на IKur и IAch. Внедрение этого препарата могло бы способствовать решению вопроса о фармакологическом лечении мерцательной аритмии.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}