
Клинический случай. Мужчина 45 лет был госпитализирован с жалобами на внезапно возникающие приступы давящей боли за грудиной, сопровождающиеся выраженной общей слабостью, головокружением, предобморочными состояниями.
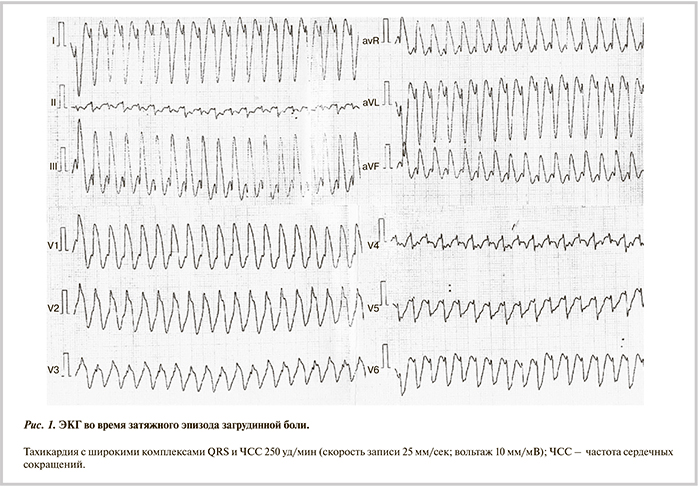
Около 2 лет назад без предшествовавшего анамнеза заболеваний сердечно-сосудистой системы (ССС) спустя 15 мин после интенсивной физической нагрузки впервые возник описанный выше приступ загрудинной боли, продолжавшийся более 0,5 ч. Вызвана бригада скорой медицинской помощи. На электрокардиограмме (ЭКГ) зарегистрирован пароксизм тахикардии с широкими комплексами QRS (рис. 1). Пациент был госпитализирован в реанимационное отделение районной больницы по месту жительства, где синусовый ритм был восстановлен посредством электроимпульсной терапии. По результатам обследования диагноз инфаркта миокарда был исключен. При эхокардиографии (ЭхоКГ) на основании утолщения межжелудочковой перегородки (МЖП) до 1,6 см диагностирована гипертрофическая необструктивная кардиомиопатия. Назначено лечение бисопрололом 5 мг/сут и фозиноприлом 10 мг/сут.
В дальнейшем у больного дважды (во время еды и в покое лежа) повторились аналогичные приступы загрудинной боли. В обоих случаях на ЭКГ была зарегистрирована тахикардия с широкими комплексами QRS, синусовый ритм был восстановлен посредством электроимпульсной терапии на догоспитальном этапе.
Кроме того, около 2 лет назад больной отметил появление общей слабости, обильную потливость и снижение переносимости привычных для него ранее физических нагрузок. Выраженность этих клинических проявлений с течением времени не усугублялась.
В раннем детском возрасте пациент перенес тяжелую инфекцию, предположительно корь, осложнившуюся правосторонним парезом лицевого нерва. Семейный анамнез больного заболеваниями системы кровообращения не отягощен.
Состояние больного при поступлении было удовлетворительным. При осмотре отмечены незначительная гипомимия лица и небольшой двусторонний асимметричный птоз. Обращала на себя внимание избыточная влажность кожных покровов. Пациент был в сознании, ориентирован, на вопросы отвечал с небольшой задержкой, при этом тяжелые когнитивные дефекты и грубые нарушения в неврологическом статусе отсутствовали. В легких выслушивалось везикулярное дыхание без хрипов. Частота сердечных сокращений (ЧСС) 86 уд/мин, артериальное давление 130/70 мм рт.ст. На ЭКГ зарегистрированы синусовая тахикардия с ЧСС 95 уд/мин, признаки гипертрофии левого желудочка (ГЛЖ), замедление предсердно-желудочкового (PQ 220 мс) и внутрижелудочкового (QRS 120 мс) проведения, одиночные желудочковые экстрасистолы. При суточном мониторировании ЭКГ по Холтеру отмечена редкая мономорфная желудочковая экстрасистолия.
По данным рентгенографии легких, а также в общих анализах крови и мочи и в коагулограмме патологические изменения отсутствовали. Сывороточные концентрации тиреотропного гормона и свободного тироксина, а также мозгового натрийуретического пептида были в пределах нормы. Анализы крови на вирус иммунодефицита человека, вирусные гепатиты и сифилис дали отрицательный результат.
В биохимическом анализе крови отмечено повышение содержания глюкозы до 6,4 ммоль/л, активности аланинаминотрансферазы (75,0 ед/л при норме 3,0—40,0 ед/л), γ-глутамилтрансферазы (97,0 ед/л при норме 10,0—64,0 ед/л), креатинфосфокиназы (КФК 347,00 ед/л при норме 15,00—200,00 ед/л), при этом активность кардиоспецифичной фракции MB КФК и концентрация сердечного тропонина I были в пределах нормы. Были также отмечены умеренные гипертриглицеридемия (3,15 ммоль/л; норма 0,50—2,30 ммоль/л) и гиперлипидемия (общий холестерин — ОХС 6,50 ммоль/л; ХС липопротеидов низкой плотности – ЛНП 3,84 ммоль/л; ХС липопротеидов высокой плотности – ЛВП 1,23 ммоль/л.
По результатам теста на толерантность к глюкозе отмечены начальные нарушения углеводного обмена. Содержание гликированного гемоглобина было в пределах нормы.
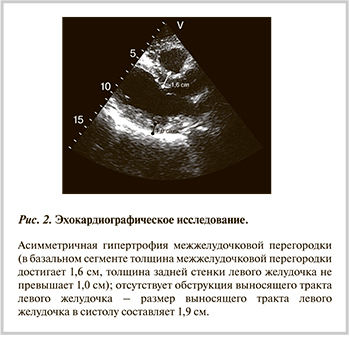
При ЭхоКГ отмечена выраженная асимметричная ГЛЖ, достигавшая диагностических критериев гипертрофической кардио-миопатии базального отдела МЖП (до 1,6 см) без признаков обструкции выносящего тракта (рис. 2). Отмечено незначительное увеличение левого предсердия до 4,2 см. Размеры других камер сердца были в пределах нормы, клапанный аппарат интактен, фракция выброса 60%, однако обращала на себя внимание повышенная эхогенность субэндокардиального слоя в проекции заднебоковой стенки ЛЖ с ограниченным участком незначительной гипокинезии. Проба на выявление скрытой коронарной недостаточности методом стресс-ЭхоКГ не доведена до субмаксимальной ЧСС из-за усталости пациента. Исходно и на максимуме нагрузки (при ЧСС 135 уд/мин) четких зон нарушения локальной сократимости левого желудочка (ЛЖ), динамики сегмента ST не отмечалось.
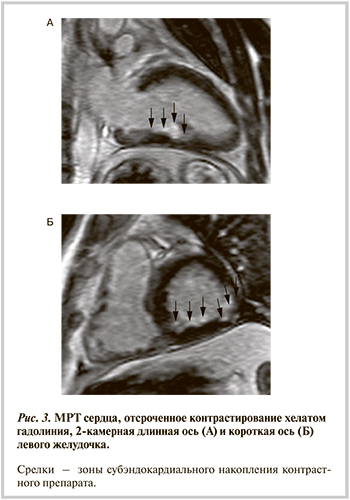
По результатам магнитно-резонансной томографии (МРТ) выявлена умеренно выраженная гипертрофия миокарда базального сегмента МЖП (до 14 мм), толщина миокарда ЛЖ в других сегментах была в пределах нормы. Толщина миокарда правого желудочка (ПЖ) составила 3—4 мм. При кино-МРТ выявлена гипокинезия боковой стенки в базальном и среднем сегментах, а также гипокинезия среднего сегмента нижней стенки. Глобальная сократимость ЛЖ была удовлетворительной. При отсроченном исследовании с контрастированием хелатом гадолиния определялось патологическое субэндокардиальное (до 60%) накопление контрастного препарата в миокарде боковой и нижней стенок ЛЖ в базальном и среднем сегментах, а также в нижних отделах базального сегмента МЖП в среднем и базальном сегментах. Накопление контрастного препарата свидетельствовало о наличии зон фиброза, а субэндокардиальный характер накопления был типичен для ишемической природы выявленных изменений (рис. 3).
Принимая во внимание пароксизмы тахикардии с широкими комплексами QRS, жалобы на боли в грудной клетке и одышку, имеющиеся факторы риска развития ишемической болезни сердца (ИБС), выявление при МРТ зон фиброза миокарда ЛЖ, позволяющие предположить их ишемический генез, пациенту провели диагностическую коронарографию. По результатам исследования коронарные артерии имеют четкие, ровные контуры, без признаков стеноза.
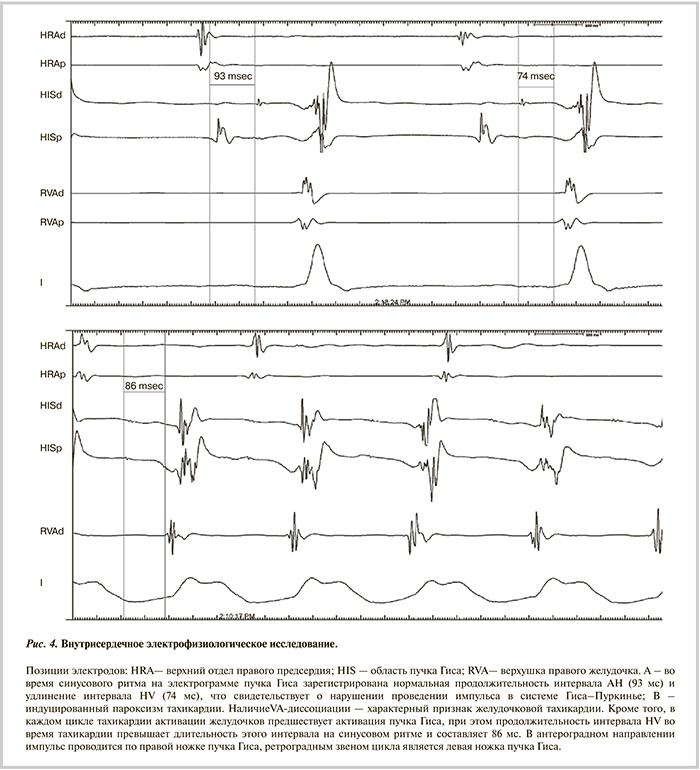
В целях индукции имевшейся у пациента аритмии и верификации диагноза проведено внутрисердечное электрофизиологическое исследование (ЭФИ). При регистрации электрограммы пучка Гиса отмечено удлинение интервала HV до 74 мс, свидетельствовующее о нарушении проведения импульса в системе Гиса—Пурикнье (рис. 4 А).
При программной стимуляции верхушки ПЖ одиночным экстрастимулом многократно индуцированы устойчивые пароксизмы тахикардии с широкими комплексами QRS (170 мс) по типу блокады левой ножки пучка Гиса с ЧСС 250 уд/мин, сопровождавшиеся головокружением, слабостью, купированные сверхчастой стимуляцией желудочков. Во время тахикардии с длительностью интервала HV 86 мс зарегистрированы признаки желудочково-предсердной диссоциации. Характер активации ствола и правой ножки пучка Гиса во время аритмии позволял сделать вывод, что наиболее вероятен диагноз пароксизмальной фасцикулярной желудочковой тахикардии (ЖТ) — циркуляция импульса в системе Гиса—Пуркинье по направлению: левая ножка пучка Гиса — общий ствол пучка Гиса — правая ножка пучка Гиса (рис. 4Б).
 При оценке неврологического статуса пациент несколько заторможен, отмечены симметричный полуптоз, снижение силы щечных мышц и силы дистальных отделов рук и ног. Походка со степпажем слева. Гипестезия на руках и ногах по полиневритическому типу. Акрогипотермия, акроцианоз, акрогипергидроз. Отмечалась тоническая судорога в мышцах кистей после физической нагрузки.
При оценке неврологического статуса пациент несколько заторможен, отмечены симметричный полуптоз, снижение силы щечных мышц и силы дистальных отделов рук и ног. Походка со степпажем слева. Гипестезия на руках и ногах по полиневритическому типу. Акрогипотермия, акроцианоз, акрогипергидроз. Отмечалась тоническая судорога в мышцах кистей после физической нагрузки.
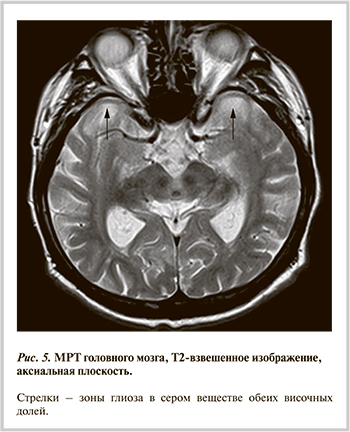
В связи с общемозговой симптоматикой и когнитивными нарушениями больному проведена МРТ головного мозга, по результатам которой выявлены множественные очаги в белом веществе головного мозга сосудистого генеза. Обращало на себя внимание наличие в передних отделах обеих височных долей зон повышенной интенсивности МР-сигнала неправильной формы с нечеткими неровными контурами, размерами 31×21 мм справа и 36×20 мм слева, которые были расценены как зоны глиоза (рис. 5). Кроме того, были выявлены признаки умеренно выраженной открытой внутренней гидроцефалии в виде расширения боковых желудочков мозга.
По данным МР-ангиографии интракраниальных артерий окклюзий, аневризм и мальформаций не выявлено, виллизиев круг замкнут. Поскольку множественные очаги сосудистого генеза в белом веществе головного мозга наиболее характерны для гипертонической болезни, признаки которой при физическом исследовании осмотре отсутствовали, пациент был направлен на консультацию офтальмолога. Выявлена начальная катаракта обоих глаз. При осмотре глазного дна отмечены сужение и неравномерное утолщение стенок артериол; диски зрительных нервов бледные, с четкими границами.
Принимая во внимание выявленное структурное заболевание сердца, имевшуюся у пациента блокаду левой ножки пучка Гиса, удлинение интервала HV до 74 мс и относительную «легкость» купирования фасцикулярной ЖТ посредством сверхчастой стимуляции ПЖ во время ЭФИ, было принято решение имплантировать пациенту кардиовертер-дефибриллятор (КД) и воздержаться от проведения радиочастотной аблации (РЧА) правой ножки пучка Гиса. Хирургическое вмешательство и послеоперационный период прошли без особенностей. Проведение РЧА было рекомендовано в случае неэффективности купирующей стимуляции и частых разрядов имплантируемого КД в дальнейшем. Начатая на догоспитальном этапе терапия бисопрололом 5 мг/сут и фозиноприлом 10 мг/сут была оставлена без изменений. Следует отметить, что непосредственная причина имевшихся у пациента ГЛЖ, нарушений ритма и проводимости сердца оставалась неясной. За больным было продолжено амбулаторное наблюдение.
С учетом клинических проявлений полиневропатии и миопатии для дальнейшего обследования и верификации диагноза пациент был направлен в Центр миастении и нейромышечных заболеваний НИИ неврологии РАМН. На основании данных объективного осмотра, клинической картины заболевания, имевшихся изменений в неврологическом статусе предположен диагноз миотонической дистрофии (МД). Результаты электромиографии подтвердили данное предположение. При проведении генетического анализа верифицирован диагноз МД I типа. Больному был назначен курс внутримышечных инъекций мексидола в целях снижения мышечного тонуса, начата терапия фенитоином в дозе 300 мг/сут.
Во время последующего наблюдения у пациента отмечено значительное прогрессирование клинических проявлений нейромышечного заболевания (нарастание мышечной слабости, утомляемости). По данным ЭКГ и ЭхоКГ, признаков прогрессирования поражения сердца при этом не наблюдалось. Спустя 3 мес у больного развился кратковременный эпизод загрудинной боли, купированный разрядом имплантированного дефибриллятора.
При телеметрическом контроле устройства документирован пароксизм ЖТ, попытки купирования которого посредством сверхчастой стимуляции были неэффективными. КД был перепрограммирован на более высокую частоту залповой антитахикардитической стимуляции. За пациентом продолжено амбулаторное наблюдение с рекомендацией повторной госпитализации для проведения РЧА очага фасцикулярной ЖТ в случае частых разрядов дефибриллятора в дальнейшем, однако в течение последующих 9 мес пароксизмы ЖТ не рецидивировали.
 При телеметрическом контроле имплантируемого КД у пациента были выявлены также множественные продолжительные пароксизмы мерцательной аритмии (МА), большинство которых были бессимптомными. Принимая во внимание высокий риск развития тромбоэмболических осложнений у больных гипертрофической кардиомиопатией и наличие зон глиоза неясной этиологии в сером веществе обеих височных долей, пациенту начали антикоагулянтную терапию ривароксабаном в дозе 20 мг/сут. Учитывая отсутствие явных клинических проявлений, относительно невысокую частоту сокращений желудочков во время пароксизмов, органическое поражение сердца и сопутствующие нарушения проводимости, приняли решение воздержаться от поддерживающей антиаритмической терапии.
При телеметрическом контроле имплантируемого КД у пациента были выявлены также множественные продолжительные пароксизмы мерцательной аритмии (МА), большинство которых были бессимптомными. Принимая во внимание высокий риск развития тромбоэмболических осложнений у больных гипертрофической кардиомиопатией и наличие зон глиоза неясной этиологии в сером веществе обеих височных долей, пациенту начали антикоагулянтную терапию ривароксабаном в дозе 20 мг/сут. Учитывая отсутствие явных клинических проявлений, относительно невысокую частоту сокращений желудочков во время пароксизмов, органическое поражение сердца и сопутствующие нарушения проводимости, приняли решение воздержаться от поддерживающей антиаритмической терапии.
МД — наиболее частая форма мышечной дистрофии взрослых (в общей популяции распространенность составляет около 1:8000). МД является наследственным заболеванием с аутосомно-доминантным типом наследования, практически 100% пенетрантностью и большим полиморфизмом клинических проявлений. Характерные симптомы заболевания — повышенный мышечный тонус и прогрессирующая слабость, обусловленные нарушением расслабления и атрофией скелетных мышц. Помимо поражения поперечнополосатых мышц характерно вовлечение других органов и систем — сердца, глаз, головного мозга, эндокринных и других органов — клинические проявления которых могут быть существенно выражены и снижать качество и продолжительность жизни пациентов [1, 2].
В настоящее время выделяют две формы заболевания. Клинические проявления МД I типа (МД1) описаны более 100 лет назад Штейнертом (Steinert). В отечественной литературе данное заболевание описано как мышечная дистрофия Россолимо—Куршмана—Штейнерта—Баттона. Более легкая форма болезни, МД II типа (МД2), описана как самостоятельное заболевание в 1994 г. [3].
Молекулярная генетика. МД1 обусловлена накоплением тринуклеотидных повторов CTG в нетранслируемой области гена протеинкиназы миотонической дистрофии (DMPK), локализованного на хромосоме 19q 13.3. У здоровых людей число повторов CTG не превышает 34, у больных DM1 это количество может составлять несколько сотен и даже тысяч. Как правило, большее число повторов CTG приводит к более тяжелым клиническим проявлениям заболевания [4].
Нестабильность протяженных повторов CTG при быстром делении клеток на ранних стадиях эмбрионального развития может обусловливать мозаичность клинических проявлений заболевания, т.е. преобладание поражений какой-либо системы органов над другими симптомами.
МД1 наследуется почти исключительно по материнской линии. Данное явление обусловлено существенно большей стабильностью и тенденцией к удлинению повторов CTG во время овогенеза, чем во время сперматогенеза Нестабильность повторов CTG обусловливает и феномен «генетической антиципации» — пациенты с тяжелыми клиническими проявлениями заболевания чаще имеют очень протяженные повторы CTG, которые претерпевают спонтанное укорочение во время гаметогенеза, и дети больных с тяжелой формой заболевания наследуют меньшее число повторов CTG, что приводит к более легким клиническим проявлениям заболевания. У больных малосимптомной формой заболевания, наоборот, чаще обнаруживают относительно небольшое число повторов CTG. Эти последовательности отличаются большей стабильностью и склонностью к удлинению в ходе гаметогенеза, что приводит к более протяженным последовательностям повторов CTG и, соответственно, большей тяжести клинических проявлений у детей.
Следует отметить, что наследственный характер заболевания у рассматриваемого пациента не прослеживался. У брата больного также отмечались неврологические симптомы, однако при обследовании в Центре миастении и нейромышечных заболеваний НИИ неврологии РАМН клинические проявления МД1 у него выявлены не были.
МД2 обусловлена накоплением избыточного количества тетрануклеотидных повторов CCTG в нетранслируемой области гена 9-го белка цинкового пальца (zincfingerprotein 9) CNF9, локализованного на хромосоме 3q 21.3. У здоровых людей число повторов CCTG в этом гене не превышает 26. У больных МД2 в среднем обнаруживают около 5000 тетрануклеотидных повторов (от 75 до 11 000). Следует отметить, что в отличие от DM1 у больных МД2 отсутствует четкая связь большего количества тетрануклеотидных повторов с более ранним возникновением и прогрессированием клинических проявлений заболевания [5].
Патогенез. МД является уникальным примером механизма РНК-токсичности, схожего при обеих формах заболевания, несмотря на то что функция конечных белковых продуктов генов DMPK и CNF9 существенно различается. Транскрипция генов DMPK и CNF9, содержащих множественные повторы CTG и CCTG, приводит к образованию мутантной РНК, которая содержит множественные повторы CUG и CCUG. Молекулы мутантной РНК связываются с белками семейств MBNL (muscleblind-like) и CELF (CUG-BP- and ETR-3-like-factors) и формируют молекулярные комплексы, напоминающие по своей пространственной структуре шпильки, накопление которых в ядрах клеток приводит к нарушениям экспрессии генетического материала. Примечательно, что отмечается нарушение экспрессии генов, локализованных на других хромосомах (т.н. транс-эффект):
- BIN1 (bridging integrator 1; нарушение экспрессии данного гена в экспериментальных исследованиях приводит к разобщению процессов возбуждения и сокращения и прогрессирующей мышечной слабости);
- хлорного канала поперечнополосатых и гладких мышц (обусловливает повышение мышечного тонуса);
- инсулинового рецептора (дисфункция приводит к инсулинорезистентности, гиперинсулинемии, нарушению толерантности к глюкозе и сахарному диабету);
- сердечного тропонина T (может приводить к развитию как гипертрофической, так и дилатационной кардиомиопатии у больных DM);
- фактора транскрипции Nkx2.5 (нарушения экспрессии Nkx2.5 приводят к нарушению синтеза белков, необходимых для нормального функционирования проводящей системы сердца; нарушения регуляции Nkx2.5 описаны у ряда больных, имеющих прогрессирующее поражение проводящей системы сердца) [6].
Клинические проявления МД. Клинические проявления МД1 и МД2 часто существенно различаются (см. таблицу). В целом МД1 является более тяжелым заболеванием. В клинической картине обычно преобладают симптомы поражения скелетных мышц — миотонический (выраженное повышение мышечного тонуса, приводящее к нарушению расслабления мышц — больные не могут быстро расслабить сжатый кулак, что часто проявляется при рукопожатиях или езде в общественном транспорте) и миопатический синдромы (появление и прогрессирование слабости различных групп мышц). Для данного заболевания характерно поражение других органов и систем, в т.ч. ССС (подробно рассмотрены ниже), которые могут быть очень выраженными. Важнейший клинический признак МД — развитие субкапсулярной катаракты в молодом возрасте (до 50 лет). Все больные, перенесшие операцию протезирования хрусталика в возрасте моложе 50 лет, должны быть обследованы для исключения МД. Клинические признаки болезни проявляются чаще всего в возрасте старше 30—40 лет. Симптомы заболевания обычно прогрессируют относительно медленно, однако продолжительность жизни больных снижена и редко превышает 60 лет. Основные причины летального исхода — развитие дыхательной недостаточности вследствие прогрессирования слабости дыхательных мышц и внезапная сердечная смерть (ВСС) [7].
Основным клиническим проявлением МД2 служит боль в мышцах различной локализации, интенсивность которой в некоторых случаях приводит к существенному ограничению двигательной активности и инвалидизации больных. Клинические проявления миотонического синдрома при этом обычно отсутствуют или выражены очень слабо. Еще один характерный симптом — слабость бедренных мышц, затрудняющая подъем по ступенькам. В целом МД2 — более доброкачественное заболевание. Выраженная слабость скелетных мышц, приводящая к ограничению двигательной активности и развитию дыхательной недостаточности, при этом тяжелые поражения сердца и других систем органов обычно отсутствуют, а средняя продолжительность жизни больных не отличается от общей популяции. Тем не менее описаны случаи тяжелого течения заболевания, обусловленного выраженной мышечной слабостью, а также случаи ВСС вследствие прогрессирования нарушений проводимости и желудочковых тахиаритмий [7].
Поражения ССС у больных МД. Поражение сердца развивается у 50—65% больных МД и чаще всего проявляется различными формами нарушений ритма и проводимости. Поражение сократительного миокарда может проявляться как развитием гипертрофии, так и проявлениями дилатационной кардиомиопатии. Характерны нарушения систолической и диастолической функций миокарда ЛЖ.
В очень редких случаях у больных МД1 при ЭхоКГ обнаруживают нарушения локальной сократимости миокарда (в основном по типу гипокинезии, чаще всего локализующиеся в области верхушки ЛЖ) [1].
При наблюдении за 361 пациентом с диагностированной МД1 в течение 10 лет смертность была в 7,3 раза выше, чем в соответствующей им по возрасту общей популяции, при этом средняя продолжительность жизни составила 53 года. Кроме того, была отмечена корреляция между возникновением клинических проявлений заболевания в раннем возрасте и летальностью. Самыми частыми причинами смерти были дыхательная недостаточность вследствие прогрессирования мышечной слабости (40%) и поражения ССС (30%). По данным различных исследований, от 2 до 30% больных МД1 умирают внезапно вследствие прогрессирования нарушений проводимости и развития полной атриовентрикулярной блокады или возникновения желудочковых аритмий (ЖА) [2].
Патофизиология. Непосредственная причина и механизмы, приводящие к поражению сердца у больных МД, остаются неизвестными. Наиболее вероятно, поражения сердца обусловлены нарушениями экспрессии генов кардиоспецифичного тропонина Т (гипертрофия миокарда и сердечная недостаточность) и фактора транскрипции Nkx2.5 (прогрессирующие нарушения проводимости) [6].
 Поскольку у больных МД1 нарушены структура и функции протеинкиназы миотонической дистрофии, которая участвует в метаболизме мышечных клеток, предположена роль метаболических нарушений. При изучении метаболизма глюкозы в кардиомиоцитах у взрослых пациентов с МД1 описано замедление утилизации глюкозы и темпов ее фосфорилирования, при этом у больных отсутствовали какие-либо изменения в кровоснабжении миокарда. Нарушения метаболизма глюкозы могут приводить к повреждению и гибели кардиомиоцитов, последующему замещению их соединительной и жировой тканями. Наличие очагов фиброза является предрасполагающим фактором к возникновению аритмий по механизму re-entry. Кроме того, сами нарушения метаболизма глюкозы могут быть причиной триггерной активности кардиомиоцитов [7].
Поскольку у больных МД1 нарушены структура и функции протеинкиназы миотонической дистрофии, которая участвует в метаболизме мышечных клеток, предположена роль метаболических нарушений. При изучении метаболизма глюкозы в кардиомиоцитах у взрослых пациентов с МД1 описано замедление утилизации глюкозы и темпов ее фосфорилирования, при этом у больных отсутствовали какие-либо изменения в кровоснабжении миокарда. Нарушения метаболизма глюкозы могут приводить к повреждению и гибели кардиомиоцитов, последующему замещению их соединительной и жировой тканями. Наличие очагов фиброза является предрасполагающим фактором к возникновению аритмий по механизму re-entry. Кроме того, сами нарушения метаболизма глюкозы могут быть причиной триггерной активности кардиомиоцитов [7].
При гистологическом исследовании образцов тканей сердца у больных МД описаны неспецифические изменения различной степени, включающие фиброз, жировую инфильтрацию, гипертрофию кардиомиоцитов и признаки фокального миокардита. При этом наиболее часто авторы отмечают преобладание распространенного поражения различных отделов проводящей системы сердца над поражением сократительного миокарда [2].
Клинические проявления поражения сердца у больных МД. У большинства больных МД1 первым клиническим проявлением служат симптомы нейромышечного заболевания (повышение мышечного тонуса, общая слабость, утомляемость). Чаще всего поражения ССС развиваются позднее, по мере прогрессирования заболевания. Изредка поражение сердца может оказаться первым проявлением заболевания. Необходимо также упомянуть о снижении когнитивных функций у больных МД1, из-за которых пациенты могут не придавать существенного значения имеющимся у них симптомам нейромышечного заболевания. У представленного пациента тяжелые клинические проявления поражения сердца преобладали над выраженностью симптомов нейромышечного заболевания.
Поражения сердца у больных МД1 могут проявляться различными формами нарушений ритма и проводимости, кардиомиопатией, а также клиническими проявлениями ИБС. Нарушения ритма и проводимости сердца отмечаются существенно чаще. В отличие большинства нейромышечных заболеваний клинические проявления сердечной недостаточности при МД1 развиваются относительно редко [2].
Распространенность и клинические проявления поражений ССС у больных МД2 изучены недостаточно хорошо. Имеются данные о том, что у больных МД2 поражение сердца отмечается реже, обычно оказывается менее тяжелым и прогрессирует медленнее, чем у больных МД1. Тем не менее при данной форме заболевания также описаны случаи опасных для жизни нарушений ритма и проводимости сердца и ВСС, вот почему все больные МД независимо от формы заболевания и тяжести клинических проявлений должны находиться под наблюдением кардиолога [7].
Нарушения проводимости сердца — самая распространенная форма поражения сердца у больных МД. Описаны поражения всех отделов проводящей системы сердца, но чаще всего в патологический процесс вовлечена система Гиса—Пуркинье, что приводит к нарушению атриовентрикулярного и внутрижелудочкового проведения. Клинически незначимые нарушения проводимости часто регистрируют на ЭКГ уже на ранних стадиях заболевания. Увеличение продолжительности интервала PQ регистрируют у 20—40% больных МД1, а расширение комплекса QRS — у 5—25% [2].
Для большинства больных МД1 характерно относительно медленное усугубление поражения проводящей системы сердца, однако описаны случаи быстрого прогрессирования блокад. Прогрессирование нарушений проводимости зачастую сопровождается такими симптомами, как одышка, головокружения, синкопе и является наиболее частой причиной ВСС этих больных [2]. Учитывая трудно прогнозируемые темпы прогрессирования заболевания, неблагоприятный прогноз и высокий риск ВСС, следует соблюдать большую осторожность в отношении нарушений проводимости у больных МД и рекомендовать незамедлительную имплантацию электрокардиостимулятора (ЭКС) в случае появления клинических проявлений, которые могут быть обусловлены прогрессирующей блокадой сердца [8].
У рассматриваемого пациента на ЭКГ отмечено незначительное замедление атриовентрикулярной (PQ 220 мс) и внутрижелудочковой (QRS 120 мс) проводимости, при этом выявленное в ходе внутрисердечного ЭФИ увеличение продолжительности интервала HV до 74 мс свидетельствовало о нарушении проведения импульса в системе Гиса—Пуркинье и указывало на целесообразность имплантации пациенту ЭКС.
Тахиаритмии. Различные формы наджелудочковых тахиаритмий достаточно часто обнаруживают при регистрации ЭКГ и суточном мониторировании ЭКГ по Холтеру. Наиболее распространенными являются МА и трепетание предсердий, обнаруживаемые у 25% больных МД1. Следует отметить, что продолжительность эпизодов МА и трепетания предсердий может варьироваться от нескольких секунд (неустойчивые пробежки) до многих часов и суток. Тактика антикоагулянтной терапии и стратификация риска развития ишемического инсульта у больных МД1 в настоящее время не разработаны, рекомендовано лечение в соответствии со стандартными схемами с использованием шкал CHADS2 и CHA2DS2-VASc [2].
В нашем случае пароксизмы МА выявлены при телеметрическом контроле ЭКС, поскольку они протекали преимущественно бессимптомно. В связи с наличием гипертрофической кардиомиопатии они имели существенное клиническое значение и обусловливали необходимость назначения антикоагулянтной терапии в связи с высоким риском развития тромбоэмболических осложнений, несмотря на отсутствие факторов риска (0 баллов по шкале CHA2DS2-VASc).
ЖА также часто обнаруживают у больных МД, однако точная распространенность их неизвестна. Описаны клинические случаи спонтанного возникновения устойчивой как мономорфной, так и полиморфной ЖТ, и даже фибрилляции желудочков (ФЖ). Всем больным МД с клиническими проявлениями, которые могут быть обусловлены ЖТ/ФЖ, рекомендовано проведение внутрисердечного ЭФИ для индукции возможных ЖА. По данным одного исследования, у 18% больных МД1 без клинических проявлений ЖТ в отсутствие клинически значимых ЖА, по данным суточного мониторирования ЭКГ по Холтеру, направленных на ЭФИ для оценки имевшихся нарушений проводимости и определения показаний к имплантации ЭКС, были индуцированы устойчивые пароксизмы ЖТ. Наиболее частой формой ЖТ, индуцируемой у больных МД в ходе ЭФИ, являются неустойчивые пробежки полиморфной ЖТ, которые в других случаях чаще всего расцениваются как диагностически незначимые неспецифические реакции. Реже отмечается индукция устойчивых пароксизмов полиморфной ЖТ, ФЖ, неустойчивых пробежек или устойчивых пароксизмов мономорфной ЖТ. В настоящее время неизвестна прогностическая значимость индукции ЖА при проведении ЭФИ больным МД без клинических проявлений и указаний на ЖТ в анамнезе. Учитывая высокий риск ВСС больных этой категории, а также сохраняющийся риск ВСС, несмотря на имплантацию ЭКС (по данным проспективного наблюдения D. Bhakta и соавт., 33,3% больных МД1 с имплантированными ЭКС умерли внезапно [9]), даже неустойчивые пробежки, в тч. полиморфной ЖТ, в ходе ЭФИ должны быть учтены при стратификации риска у больных и определении дальнейшей тактики лечения [2].
Мономорфная ЖТ также часто обнаруживается у больных МД1. Возможные механизмы возникновения включают re-entry между ножками пучка Гиса (т.н. фасцикулярная ЖТ — bundle branch re-entry; наиболее типичный вариант, при котором одна из ножек пучка Гиса является антероградным звеном цикла re-entry, а другая — ретроградным), re-entry вокруг очагов жирового и фиброзного замещения миокарда (возможный вариант) и триггерную активность (редко). Большая распространенность ЖТ, обусловленной циркуляцией импульса между ножками пучка Гиса, может быть объяснена преимущественным поражением системы Гиса—Пуркинье, что приводит к неравномерному замедлению проведения импульса и возникновению антероградной блокады проведения по одной из ножек при сохранении проведения в ретроградном направлении, что является необходимым условием для замыкания петли re-entry. Интерфасцикулярная ЖТ, возникающая вследствие циркуляции возбуждения между ветвями левой ножки пучка Гиса, также описана у больных МД1. Индукция ЖТ с участием ножек пучка Гиса и интрафасцикулярной ЖТ в ходе ЭФИ часто затруднена. Запуск тахикардии может потребовать специальных, более агрессивных протоколов стимуляции (например, последовательность короткого-длинного-короткого интервалов сцепления экстрастимулов), которые должны быть выполнены не только из верхушки и выносящего тракта ПЖ, но также из предсердий [2].
Верификация ЖТ с участием ножек пучка Гиса и интерфасцикулярной ЖТ у больных МД1 имеет существенное клиническое значение, поскольку данные формы аритмии могут быть успешно устранены посредством радиочастотной катетерной аблации правой ножки пучка Гиса или одной из ветвей левой ножки пучка Гиса соответственно. Следует отметить, что эффективная РЧА не полностью устраняет риск ВСС у больных DM и не избавляет их от необходимости имплантации КД, поскольку примерно в 25% случаев данные формы аритмии сопровождаются также устойчивыми пароксизмами ЖТ, исходящими из очагов фиброзного и жирового замещения сократительного миокарда [10].
У представленного пациента отмечались пароксизмы фасцикулярной ЖТ. Принимая во внимание относительно редкие пароксизмы, сопутствующие нарушения проводимости, легкость купирования аритмии посредством «залповой» стимуляции во время ЭФИ, а также отрицательное прогностическое значение полной блокады левой ножки пучка Гиса, было принято решение воздержаться от проведения РЧА правой ножки пучка Гиса.


Сердечная недостаточность. Тяжелую дисфункцию миокарда («миотоническую кардиомиопатию») редко отмечают у больных МД (обычно у пациентов с тяжелыми клиническими проявлениями нейромышечного заболевания), при этом признаки субклинического поражения миокарда обнаруживают достаточно часто. Клинические проявления сердечной недостаточности как таковой у пациентов обычно отсутствуют в связи с ограничением двигательной активности из-за выраженных нейромышечных симптомов. Кроме того, в связи с когнитивной дисфункцией больные МД1 могут не придавать существенного значения имеющимся у них симптомам. При МД1 возможно развитие как дилатационной, так и гипертрофической кардиомиопатии, описаны редкие случаи избыточной трабекулярности миокарда ЛЖ [2].
При проведении трансторакальной ЭхоКГ у 382 больных МД1 были выявлены следующие структурные изменения сердца: гипертрофия миокарда ЛЖ (20%), дилатация ЛЖ (19%), снижение фракции выброса ЛЖ (14%), нарушения локальной сократимости миокарда ЛЖ (11%), расширение левого предсердия (6%) [11]. При ЭхоКГ у 25—40% больных МД1 отмечают пролапс митрального клапана. Нарушение диастолической функции миокарда ЛЖ некоторые авторы считают эквивалентом миотонии скелетных мышц; у многих пациентов признаки диастолической дисфункции по результатам тканевой миокардиальной допплерографии обнаруживают до появления иных эхокардиографических признаков поражения сердца [2].
При проведении МРТ сердца больным МД1 также часто обнаруживают признаки структурного поражения сердца. Так, по данным исследования M.C. Hermans и соавт., в ходе которого 80 больным МД1 было проведено МРТ сердца с контрастированием хелатом гадолиния, структурные и функциональные патологические изменения были выявлены у 44% пациентов, среди которых отмечены: снижение фракции выброса ЛЖ (25%), дилатация ЛЖ (9%), гипертрофия миокарда ЛЖ (8%), снижение фракции выброса ПЖ (5%), дилатация ПЖ (1%). Следует отметить, что вовлечение в патологический процесс миокарда ПЖ было отмечено только у больных с выраженным поражением миокарда ЛЖ и только у мужчин. Нарушения локальной сократимости миокарда ЛЖ (зоны гипокинезии) описаны у 14% больных, при этом в 4% они сочетались с локальным истончением миокарда. У 13% больных обнаружены зоны отсроченного контрастирования хелатом гадолиния, что указывает на наличие очагов фиброзного замещения миокарда ЛЖ. У всех пациентов зоны накопления контрастного препарата располагались в МЖП, нижней или боковой стенках ЛЖ.
При этом в 10% случаев описаны ограниченные интрамуральные очаги, а в 3% — обширные зоны субэндокардиального или трансмурального фиброза. Примечательно, что более тяжелые поражения сердца отмечены у мужчин, больных, имевших изменения на ЭКГ, и пациентов старшего возраста, при этом степень выраженности структурного поражения сердца не зависела от давности заболевания, числа повторов CTG и тяжести симптомов нейромышечного заболевания [12].
ИБС. Различные клинические формы ИБС (стабильная стенокардия, нестабильная стенокардия и острый инфаркт миокарда) также встречаются у больных МД1. Описаны случаи стенокардии, обусловленной нарушениями микроциркуляции у пациентов с характерными ангинозными приступами, подтвержденными положительным результатом дипиридамоловой стресс-сцинтиграфии с 202Tl, в отсутствие гемодинамически значимых стенозов по результатам коронарографии [2].
Обсуждение. Представленный клинический случай интересен как с точки зрения диагностического поиска, так и в связи с трудностями лечения пациента. Основанием для подозрения на нейромышечное заболевание и направления на консультацию невролога и дальнейшее специализированное обследование стали жалобы на повышенную утомляемость, которые пациент отмечал длительное время, но не придавал им значения, и транзиторное повышение активности КФК в биохимическом анализе крови. Ведущим клиническим проявлением заболевания стали пароксизмы ЖТ, сопровождавшиеся продолжительной болью в груди.
Фасцикулярная ЖТ является одной из наиболее тяжелых аритмий. Крайне высокая частота сокращений желудочков часто приводит к острой левожелудочковой недостаточности, аритмическому шоку, обмороку, клинической картине остановки кровообращения. Высок риск трансформации в ФЖ, которая служит непосредственной причиной ВСС. Методом выбора в лечении данной формы аритмии является РЧА одной из ножек пучка Гиса вместе с имплантацией КД, поскольку более чем у 25% больных в дальнейшем возникают другие опасные для жизни ЖА [10]. Примечательно, что у пациента отсутствовали синкопальные и пресинкопальные состояния, ни разу не происходило трансформации ЖТ в ФЖ. С учетом редких пароксизмов, нарушений внутрижелудочковой проводимости, «легкости» купирования пароксизмов ЖТ посредством частой и сверхчастой стимуляции в ходе ЭФИ, а также отрицательного прогностического значения полной блокады левой ножки пучка Гиса было принято решение воздержаться от РЧА.
Диагностированная у пациента МА, несмотря на малосимптомность, также имеет существенное клиническое значение.
В соответствии с рекомендациями Европейского общества кардиологов и Американской ассоциации сердца, учитывая высокий риск развития кардиоэмболических инсультов, всем больным гипертрофической кардиомиопатией и МА, независимо от имеющихся у них факторов риска, показан систематический прием пероральных антикоагулянтов [13—14]. Больному была начата антикоагулянтная терапия ривароксабаном 20 мг/сут.


