
Основной причиной гибели и инвалидизации больных с мерцательной аритмией (МА) являются тромбоэмболические осложнения (ТЭО). В большинстве случаев источником тромбоэмболии служит левое предсердие (ЛП). Частота развития тромбоза ЛП колеблется от 8 до 25%, причем более 90% тромбов локализованы в его ушке [1].
Предполагается, что в ряде случаев тромб ЛП не является источником тромбоэмболии. В одной небольшой работе был проанализирован иммунохимический состав тромба в предсердии и эмбола. Оказалось, что эти тромбы различны по структуре. Предсердные тромбы содержали аморфное вещество и фибрин, тогда как эмболы состояли из большого числа тромбоцитов и фибрина. Это дало возможность авторам сделать осторожный вывод: эмболы могут образовываться непосредственно в кровотоке у больных с высокой активностью свертывающей системы [2].
Следует, однако, учитывать, что имеющиеся тромбы не всегда отрываются, а отсутствие тромба не означает, что риск развития ТЭО отсутствует. Один из возможных факторов, влияющих на риск развития тромбоэмболии, — изменение уровня трансляции мРНК фактора ХШа в тромбоцитах. Известно, что в тромбоцитах содержатся мРНК и все необходимые для трансляции белки. Подавление или активация трансляции тех или иных мРНК модулируется на фоне активации тромбоцитов, взаимодействия тромбоцитарных интегринов с лигандами [3, 4]. Как известно, фактор ХIIIа способствует стабилизации фибринового тромба.
У больных с МА выявлена закономерность в уровне трансляции мРНК гена субъединицы А фактора XIII в тромбоцитах, в зависимости от того, содержатся ли эти тромбоциты в тромбе ушка ЛП или в тромбоэмболе. Самый низкий уровень трансляции мРНК характерен для тромбоцитов, находящихся в тромбоэмболе (в отсутствие тромбоза ушка ЛП). Промежуточный уровень трансляции мРНК характерен для тромбоцитов в составе тромбоэмбола, если в ушке ЛП сохраняется тромб. И наконец, самый высокий уровень трансляции мРНК субъединицы А фактора XIII определяется в тромбоцитах в составе тромба в отсутствие периферических тромбоэмболий, так как этот тромб характеризуется наибольшей прочностью [5]. Молекулярный механизм регуляции трансляции мРНК фактора XIII и, соответственно, способы влияния на него неизвестны. Тем не менее можно предполагать, что в дальнейшем это может стать перспективным направлением в создании новых антитромботических препаратов.
Кроме тромбоза ушка ЛП и его структурных особенностей риск развития ТЭО повышают и другие факторы, которые обсуждаются ниже.
Особенности функционирования системы гемостаза при МА
Первым этапом нарушений в системе гемостаза при МА считают развитие дисфункции эндотелия (ДЭ) в эндокарде ушка ЛП, хотя строгих доказательств первичности такого поражения нет. Локальные протромботические сдвиги в эндокарде могут объяснять то, что тромб гораздо чаще образуется в ушке ЛП, а не правого предсердия [6], хотя степень механической дисфункции и стаз крови в ушках одинаковы [7]. Например, в исследовании in vivo, проведенном на макаках-крабоедах, выявлено, что активность физиологической антикоагулянтной системы белка С в эндотелиальных клетках в ушке ЛП почти в 2 раза ниже, чем в ушке правого предсердия. Это связано с низким уровнем экспрессии тромбомодулина вследствие локальной ДЭ [8].
Предполагается, что локальная ДЭ в эндокарде ушка ЛП способна вызывать перестройку метаболической активности эндотелия других сосудистых областей. Генерализованные нарушения функции эндотелия приводят к повышению прокоагулянтной активности крови на фоне снижения фибринолитического резерва (табл. 1). Однако изменения на одном участке эндотелия могут быть прямо противоположными изменениям в другом. Именно поэтому окончательное суждение о системной ДЭ весьма затруднительно. Соответственно, к оценке значимости регистрируемых изменений надо подходить с большой осторожностью.
Таблица 1. Изменения уровней факторов гемостаза у больных с МА.
Примечание. ↑— повышение уровня; ↓— снижение уровня; МА — мерцательная аритмия; ФВБ — фактор Виллебранда; ИПТФ — ингибитор пути тканевого фактора; ИАП-1 — ингибитор активатора плазминогена 1-го типа.
У больных с постоянной формой МА продемонстрировано повышение в крови уровня комплекса тромбин—антитромбин III и фрагментов протромбина 1+2, особенно при клапанной форме МА [9]. У больных с МА и ишемическим инсультом по сравнению с больными с инсультом и синусовым ритмом регистрировались повышенные концентрации фактора Виллебранда (ФВБ), фактора VIII, фибриногена, β-тромбоглобулина, D-димера и тромбоцитарного фактора 4, что свидетельствует об активации тромбоцитов и свертывающей системы крови [10]. При этом отмечалось снижение фибри- нолитической активности крови вследствие повышения концентрации ингибитора активатора плазминогена 1-го типа (ИАП-1) [11]. Нарушения в системе фибринолиза косвенно подтверждают результаты работы N. Petersen и соавт., которые показали, что у больных с кардиоэмболическим инсультом существенно снижен уровень активности плазминогена [12]. В другой работе выявлено снижение уровня ИАП-1 у больных с кардиоэмболическим инсультом по сравнению с атеротромботическим [13].
Вид и степень изменений в системе гемостаза зависят от общей длительности МА. При постоянной форме МА выявлено повышение уровней ФВБ, фибриногена и растворимого P-селектина, при персистирующей — повышение уровня ФВБ и фибриногена, а при пароксизмальной — растворимого P-селектина и фибриногена [14]. Противоречивые результаты получены в других работах, где при постоянной форме МА также выявлено повышение уровней ФВБ, фибриногена и растворимого P-селектина, при пароксизмальной форме повышались уровни только фибриногена и ФВБ, а у больных с персистирующей формой МА уровни всех трех факторов были в пределах нормы [15]. S. Kamath и соавт. [16] выявили повышение уровней D-димера и β-тромбоглобулина при всех формах МА, самые высокие уровни регистрировались у больных с постоянной формой. При этом уровни фибриногена и растворимого Р-селектина не различались у больных с любой формой МА и у здоровых лиц. Не выявлено различий в концентрации растворимых Р- и Е-селектинов, ФВБ, С-реактивного белка (СРБ), интерлейкина-6 (ИЛ-6) и тканевого фактора в зависимости от длительности МА, оцененной по данным длительного мониторинга электрокардиограммы. Уровни всех этих маркеров были практически одинаковыми у больных без МА во время исследования (МА в анамнезе) и у больных с суммарной продолжительностью МА 0,1—10, 10—50 и >50% времени наблюдения [17].
Маркеры тромбоза ушка ЛП при МА
Механизм тромбообразования при МА изучен недостаточно. Имеющиеся сведения нередко противоречат друг другу и не складываются в единую картину. Механизмы и клиническое значение повышения или снижения уровней того или иного фактора системы гемостаза до сих пор не расшифрованы. Прямое измерение концентрации факторов свертывания при МА с целью изучения процессов тромбообразования в клинической практике не используется. Тем не менее выявление маркеров внутрисердечного тромбоза при МА может помочь в комплексной оценке риска развития ТЭО.
Фибриноген, фрагменты протромбина 1+2 и комплекс тромбин—антитромбин III — повышенный уровень ассоциирован с наличием спонтанного контрастирования ЛП или его ушка по данным чреспищеводной эхокардиографии (ЭхоКГ) [18].
Фибрин-мономер — чувствительный маркер гиперкоагуляции in vivo. В работе Н. Okuyama и соавт. [19] установлена ассоциация между повышением уровня фибрин-мономера с тромбозом ушка ЛП (независимо от наличия или отсутствия МА).
D-димер. В ряде работ выявлена связь между наличием тромба в ушке ЛП и высоким уровнем D-димера. Эти наблюдения подтвердились и нами при обследовании 114 больных с персистирующей формой МА. Показано, что уровень D-димера является независимым предиктором тромбоза ушка ЛП [20]; это подтверждается и другими исследованиями [21]. Уровень D-димера менее 1,15 мкг/мл может служить маркером отсутствия тромба в ушке ЛП при МА (прогностическая ценность отрицательного результата 97%). У больных с уровнем D-димера более 1,15 мкг/мл тромб ушка ЛП выявлялся в 21,8% случаев, а при уровне менее 1,15 мкг/мл — только в 3%. Противоречивые результаты получены в работе S. Sugiura и соавт. [22] — у 19 из 23 больных с тромбозом ушка ЛП на фоне МА уровень D-димера не превышал 0,5 мкг/мл.
По данным Е.С. Кропачевой и соавт. [23], изменение уровня D-димера можно использовать для оценки эффективности терапии непрямыми антикоагулянтами. После растворения тромба в ушке ЛП уровень D-димера существенно снижался.
ИАП-1. Это протеиназный ингибитор, участвующий в регуляции активности фибринолитической системы. Постоянная форма МА, как правило, сопровождается повышением его уровня. Повышенный уровень ИАП-1 наблюдался у больных, у которых тромб в ЛП не растворялся, несмотря на терапию варфарином; связь уровня ИАП-1 с образованием тромба оказалась недостоверной [24]. Изучая уровень этого фактора, мы получили довольно интересные результаты [20]. Оказалось, что при МА низкий уровень ИАП-1 является фактором риска тромбоза ушка ЛП. В исследование были включены 92 больных с длительностью МА более 48 ч. У больных с тромбом в ушке ЛП среднее давление в легочной артерии, диаметр выносящего тракта левого желудочка и уровень D-димера были выше, а фракция выброса левого желудочка и уровень ИАП-1 — ниже, чем у больных без тромба. Независимыми факторами риска тромбоза ушка ЛП оказались только низкий уровень ИАП-1 (отношение шансов — ОШ 0,51 при 95% доверительном интервале — ДИ от 0,276 до 0,936; p=0,03) и повышенный уровень D-димера (ОШ 1,01 при 95% ДИ от 1,001 до 1,014; p=0,026). Выявленное снижение уровня ИАП-1 у больных с тромбом в ушке ЛП, вероятно, указывает на то, что формирование тромба является следствием своеобразного истощения факторов фибринолиза, и, соответственно, сопровождается повышенным потреблением ИАП-1. Это согласуется с результатами V. Roldan и соавт., выявившими, что у больных с кардиоэмболическим инсультом по сравнению с атеротромботическим уровень ИАП-1 снижен; возможно, это связано с формированием тромба в ушке ЛП [25].
Активация тромбоцитарного звена гемостаза при МА, несомненно, играет роль в развитии тромбоза. Наиболее высокий риск тромбоза отмечается у больных с клапанной формой МА. У таких больных наблюдается не только активация тромбоцитов, но и их повышенное разрушение с обнаружением в кровотоке свободных фрагментов мембранных гликопротеидов Ib [26]. Доказано, что на активацию тромбоцитов, как и на частоту обнаружения внутрипредсердного тромба, влияет замедление кровотока в ушке ЛП. Вместе с тем неясно, оказывают ли прямое влияние системные нарушения гемостаза и активация тромбоцитов на формирование тромба в ушке ЛП.
Интересные результаты получены S. Willoughby и соавт., которые показали, что степень активации тромбоцитарного звена гемостаза при МА зависит от участка сосудистого русла. Уровень экспрессии Р-селектина и степень индуцированной АДФ агрегации тромбоцитов были выше в образцах крови из ЛП по сравнению с образцами из правого предсердия и бедренной вены [27]. Роль выявленных нарушений в процессе тромбообразования при МА нуждается в дальнейшем изучении.
ФВБ. Повышение уровня ФВБ в плазме увеличивает риск развития ТЭО при МА [28]. Установлено, что уровень ФВБ достоверно выше при наличии участков адгезии тромбоцитов на эндокарде или тромба в ушке ЛП [29].
Тромбомодулин и ингибитор пути тканевого фактора (ИПТФ). У больных с синусовым ритмом с помощью иммуногистохимического анализа показано, что почти вся поверхность эндотелия предсердий покрыта молекулами тромбомодулина и ИПТФ, за счет чего эндокард в норме является антикоагулянтным барьером. На экспериментальной модели пароксизмальной МА у крыс (сверхчастая электрокардиостимуляция предсердий в течение 8 ч) установлено, что МА приводит к уменьшению экспрессии генов тромбомодулина и ИПТФ (по подавлению синтеза мРНК) и, соответственно, к снижению уровня этих белков в эндокарде ЛП. В эндокарде левого желудочка экспрессия тромбомодулина и ИПТФ после сверхчастой электрокардиостимуляции не изменялась [30].
Липопротеид(а)— макромолекула, содержащая апопротеин(а). Структура молекулы апопротеина(а) гомологична структуре плазминогена. Липопротеид(а) оказывает ингибирующее действие на синтез тканевого активатора плазминогена, способен связываться с фибрином и конкурентно взаимодействовать с клеточными рецепторами плазминогена. Он также снижает активность эндогенной системы фибринолиза, что показано на экспериментальной модели венозного тромбоза [31].
Повышение уровня липопротеида(а) отмечено у больных с постоянной формой МА и наличием тромбов в ЛП [32, 33]. Пороговый уровень липопротеида(а) более 30 мг% служит независимым предиктором как наличия тромба в ушке ЛП, так и развития ТЭО [32, 33].
Воспаление как фактор риска развития тромбоза при МА
Вслед за атеросклерозом, в механизме развития которого воспалительным реакциям стали отводить одну из основных ролей, подобные закономерности все чаще находят и при МА. Проведено большое количество исследований, доказывающих важную роль воспаления в развитии МА и ее осложнений [34—38]. По крайней мере некоторые маркеры воспаления — ОРБ, ИЛ-6 и α-фактор некроза опухолей (α-ФНО) — связаны с этой аритмией.
Изучена связь маркеров воспаления и протромботических нарушений при МА [11, 39, 40]. Повышенный уровень СРБ, определенный высокочувствительным методом (вч-СРБ), сопровождался увеличением вязкости крови и уровня фибриногена, а повышенный уровень ИЛ-6 — увеличением экспрессии тканевого фактора. В небольшом исследовании ИЛ-6 оказался независимым предиктором ишемического инсульта при МА. Чем выше был уровень вч-СРБ, тем более выраженным было спонтанное контрастирование ЛП. Риск развития инсульта был наибольшим при сочетании спонтанного контрастирования, низкой скорости изгнания крови из ушка ЛП и высокого уровня вч-СРБ. На большой группе больных с МА показано, что уровень вч-СРБ является независимым предиктором инсульта; повышение уровня вч-СРБ коррелировало с увеличением риска развития инсульта, оцененного по шкале CHADS2 [11].
Определенный параллелизм прослеживается и между воспалительными маркерами и тромбозом ушка ЛП при МА. Именно из-за воспалительного повреждения эндокарда у больных с аритмиями на фоне ревматических клапанных пороков отмечаются более выраженные протромботические сдвиги. При неклапанной форме МА выявлена ассоциация между наличием тромба в ушке ЛП и повышением уровней моноцитарного хемотаксического фактора 1-го типа и молекул адгезии (VCAM-1) [41].
Тучные клетки участвуют в процессах локального воспаления, осуществляют синтез ряда вазоактивных и протромбогенных медиаторов — гистамина, гепарина, протеолитических ферментов, цитокинов, секреция которых регулируется фактором роста тучных клеток. При тромбозе ушка ЛП выявлены значительное увеличение числа этих клеток в эндокарде ушка, перемещение их в верхний слой эндокарда и повышение экспрессии фактора роста тучных клеток [42]. При этом количество макрофагов, лимфоцитов и нейтрофилов в эндокарде ушка оставалось таким же, как в группе контроля. Таким образом, скопление тучных клеток в ушке ЛП не было следствием неспецифической воспалительной реакции при МА. Известно, что активированные тучные клетки выделяют α-ФНО, повышают экспрессию молекул адгезии VCAM-1 и ICAM-1, что, возможно, приводит к формированию тромба в ушке ЛП [43].
Роль ДЭ при формировании тромба в ушке ЛП
В эндокарде ЛП выявлен высокий уровень базальной секреции оксида азота (NO) [44]. В норме в эндокарде ЛП синтезируется в 3 раза больше NO, чем в эндотелии аорты и эндокарде желудочков. Таким образом, ЛП участвует в регуляции системного сосудистого тонуса и процессов тромбообразования. МА приводит к значительному снижению экспрессии эндотелиальной NO-синтетазы и, соответственно, к падению концентрации NO в эндокарде ЛП. В эндокарде правого предсердия и в аорте экспрессия NO-синтетазы и концентрация NO не отличаются от таковых при синусовом ритме [41]. Падение экспрессии эндотелиальной NO-синтетазы в эндокарде ЛП при МА, скорее всего, обусловлено гемодинамическими нарушениями. Напряжение сдвига активирует тирозинкиназу Src, что повышает экспрессию NO-синтетазы за счет как активации транскрипции, так и повышения стабильности мРНК. Напряжение сдвига также повышает биодоступность NO в эндотелии за счет фосфорилирования NO-синтетазы. Помимо нарушений гемодинамики при МА отмечается повышение синтеза супероксидных радикалов [45, 46], которые инактивируют NO. Следствием системного снижения синтеза и уменьшения стабильности NO является снижение уровней нитритов и нитратов в плазме больного МА, а также уровня цГМФ в тромбоцитах, что приводит к протромботическим сдвигам и формированию тромба в ушке ЛП [47].
Роль механической дисфункции при формировании тромба в ушке ЛП
Ведущей причиной тромбообразования при МА является механическая дисфункция предсердий, в основе которой лежат электрическое ремоделирование, нарушение сократимости кардиомиоцитов и структурное ремоделирование стенки сердца. Электрическое ремоделирование проявляется изменением уровня внутриклеточного кальция и уменьшением продолжительности потенциала действия. Электрическое ремоделирование нарушает сократимость предсердий, что приводит к стазу крови, а позднее вызывает структурное ремоделирование [48]. Морфологической основой структурного ремоделирования служит развитие фиброза стенки предсердий. Фиброз, с одной стороны, является следствием МА, а с другой — провоцирующим фактором ее развития. Фиброз характерен для всех форм МА, включая изолированную [49—51]. В настоящее время появились неинвазивные способы выявления фиброза — магнитно-резонансная томография с отсроченным контрастированием и ЭхоКГ с использованием интегрированного обратного рассеивания [52, 53].
Патогенез фиброза предсердий при МА изучен недостаточно. Предполагается участие ренин-ангиотензин-альдостероновой системы (РААС), трансформирующего β-фактора роста и окислительного стресса [54, 55]. Ранее была доказана роль РААС в развитии фиброза миокарда желудочков при артериальной гипертонии, сердечной недостаточности, инфаркте миокарда и кардиомиопатии [54]. У больных с первичным гиперальдостеронизмом значимо увеличивается риск развития МА [56], а повышение экспрессии ангиотензина II у трансгенных мышей помимо развития аритмии сопровождается дилатацией и фиброзом предсердий [57]. Ангиотензин II стимулирует гиперпродукцию трансформирующего β-фактора роста, который индуцирует накопление в межклеточном матриксе миофибробластов, активирует синтез металлопротеиназ, усиливает воспалительную инфильтрацию и приводит к развитию фиброза [58, 59]. В экспериментах на мышах гиперпродукция трансформирующего β-фактора роста в тканях сердца сопровождалась развитием избирательного фиброза предсердий и МА [60, 61]. Особенно следует отметить, что гиперпродукция трансформирующего β-фактора роста отмечалась как в предсердиях, так и в желудочках, но фиброз развивался только в предсердиях. Причин тому может быть несколько. Во-первых, в миокарде предсердий исходно отмечается более высокое содержание фибробластов [62]. Во-вторых, в условиях in vitro и in vivoустановлено, что предсердные фибробласты активнее, чем желудочковые, превращаются в миофибробласты [63]. Наконец, в ответ на стимуляцию факторами роста в предсердных фибробластах пролиферация и транскрипция происходят значительно активнее, чем в желудочковых [63].
На клеточном уровне возможны два различных пути развития фиброза — замещение соединительной тканью погибших в результате активации апоптоза кардиомиоцитов и первичная активация фибробластов (в ответ на растяжение стенки предсердия, воспалительные и оксидантные сдвиги), приводящая к гиперпродукции внеклеточного матрикса. При МА реализуются оба механизма. Активация фибробластов при МА сопровождается гиперпродукцией матриксных металлопротеиназ [64], что стимулирует экспрессию целого ряда провоспалительных цитокинов, индуцирующих апоптоз предсердных кардиомиоцитов. У больных с МА экспрессия матриксных металлопротеиназ в ткани ушка ЛП существенно выше, чем у лиц без аритмии [65].
Основным результатом формирования фиброза при МА является развитие механической дисфункции предсердий. Соответственно можно предположить наличие связи между распространенностью фиброза и риском образования тромба в ЛП. Результатов прямого сопоставления степени распространенности фиброза предсердий с риском тромбообразования не получено. Но косвенные свидетельства такого влияния есть. Так, с помощью магнитно-резонансной томографии с отсроченным контрастированием изучена структура ЛП у 387 больных с МА. Степень распространенности фиброза оценивалась в процентах от площади ЛП. У больных с инсультом в анамнезе распространенность фиброза была достоверно выше, чем у лиц без инсульта, и составила 24,4±12,4% по сравнению с 16,2±9,9% (p<0,01). При распространенности фиброза <8,5% инсульт встречался в 2,8% случаев, а при распространенности фиброза >21,1% — в 52,8%. Аналогичные результаты получены для оценок по шкале CHADS2. У больных с оценкой 2 балла и более распространенность фиброза составила 18,7±11,4% по сравнению с 14,7±9,2% у больных с оценкой по шкале CHADS2 <2 баллов. При проведении многофакторного анализа фиброз ЛП оказался независимым предиктором инсульта. Наличие распространенного фиброза (>21,1% по сравнению с <8,5%) увеличивало относительный риск развития инсульта почти в 4 раза [66]. Однако ретроспективный характер исследования не дает достаточных оснований полагать, что степень распространенности фиброза, выявленная в момент включения в исследование, соответствует той, которая была у больного в момент развития инсульта.
В настоящий момент влияние фиброза на формирование тромба в ЛП и развитие ТЭО при МА не может считаться доказанным.
Влияние гормональных нарушений на формирование тромба в ушке ЛП
В ряде работ описана ассоциация повышенного уровня предсердного натрийуретического пептида (ПНУП) с риском развития инсульта [67, 68], с низкой скоростью изгнания крови из ушка ЛП [69] и с тромбозом ушка ЛП [67] у больных с МА. По всей видимости, повышение уровня ПНУП является только маркером высокого риска тромбоза. При этом механическая дисфункция и фиброз стенки предсердий, приводящие к гормональным нарушениям, представляют собой значимые патогенетические звенья в процессе тромбообразования при МА.
В работе Y. Okada и соавт. [70] изучена возможность использования уровня ПНУП в качестве маркера внутрисердечного тромба у больных с инсультом и МА. Актуальность поставленной задачи определяется тем, что не всем больным с инсультом из-за тяжести состояния можно выполнить чреспищеводную ЭхоКГ, а диагностика внутрисердечного тромбоза важна при выборе анти- тромботического лечения. Обследованы 67 больных с МА, госпитализированных по поводу ишемического инсульта или транзиторной ишемической атаки. При чреспищеводной ЭхоКГ тромбоз ЛП выявлен у 17 (25,4%) больных. Уровень ПНУП был достоверно выше у больных с тромбозом, чем без тромбоза, — 189,8 (141,4—473,2) пг/мл по сравнению с 117,9 (70,3—187,1) пг/мл (p=0,012). По результатам многофакторного анализа повышение уровня этого биомаркера >140 пг/мл оказалось независимым предиктором обнаружения тромба (ОШ 5,62 при 95% ДИ от 1,39 до 22,66; р=0,015), чувствительность и специфичность составляли 76,5 и 62% соответственно. Основным ограничением данной работы, не позволяющим прямо переносить полученные результаты в практику, является выборочное выполнение чреспищеводной ЭхоКГ. Возможность использования ПНУП в качестве маркера тромбоза ушка ЛП должна быть изучена в рамках крупного проспективного регистра.
Генетические аспекты повышенного риска тромбообразования и ТЭО при МА
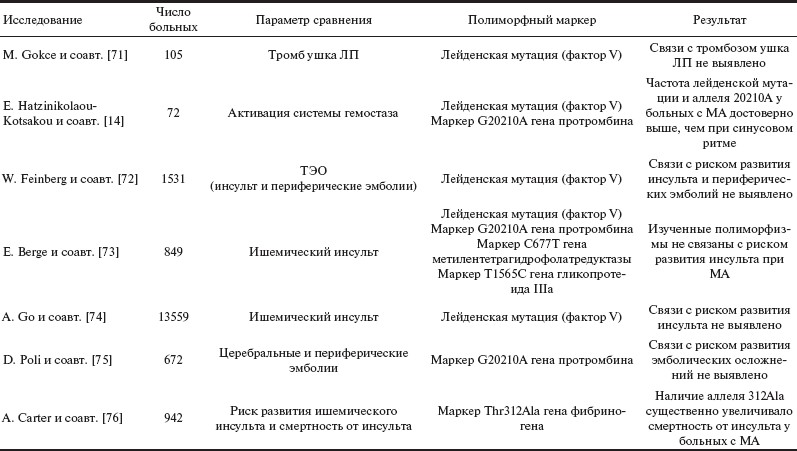
Данный вопрос изучен явно недостаточно, в табл. 2 суммированы некоторые данные, касающиеся связи генетических особенностей факторов гемостаза с тромбозом ушка ЛП или с ТЭО при МА.
Таблица 2. Связь полиморфных маркеров генов системы гемостаза с тромбозом ушка ЛП или ТЭО при МА.
Примечание. МА — мерцательная аритмия; ТЭО — тромбоэмболические осложнения; ЛП — левое предсердие.
Риск тромбоза ушка ЛП
Наиболее изученным генетическим предиктором венозного тромбоза является лейденская мутация — замена Арг на Глу в положении 506 гена фактора V. Эта мутация приводит к тому, что фактор Va становится устойчивым к расщепляющему действию активированного белка C. Изучение лейденской мутации при МА дало противоречивые результаты.
В небольшом европейском исследовании, включившем 105 больных с МА без клапанных пороков сердца, роль этой мутации как фактора риска тромбоза ушка ЛП не подтвердилась [71]. В России аналогичная работа проведена в лаборатории, возглавляемой Е.П. Панченко [77]. В исследование включены 48 больных с МА. Гетерозиготных носителей лейденской мутации было 8,3%. У всех носителей лейденской мутации выявлялся тромб в ушке ЛП. В то же время не получил пока объяснения тот факт, что частота лейденской мутации в группе существенно превысила среднестатистический уровень этого генетического дефекта в российской популяции.
Результаты нашей работы подтверждают важную роль фактора V в развитии тромбоза ЛП [78]. Изучена распространенность полиморфного маркера гена фактора V C-426T и лейденской мутации у 53 больных с персистирующей формой МА (длительность пароксизма — от 48 ч до 90 сут) в зависимости от наличия тромбоза ушка ЛП (диагностирован у 11 больных). При многофакторном анализе выявлены следующие независимые факторы риска тромбоза ушка ЛП: ишемическая болезнь сердца (ОШ 15,1 при 95% ДИ от 1,16 до 196,54; p=0,039), генотип TT полиморфного маркера C-426T (ОШ 51,2 при 95% ДИ от 1,88 до 1396,39; p=0,02) и уменьшение максимальной скорости изгнания крови из ушка ЛП менее 20 см/с (ОШ 29,17 при 95% ДИ от 2,32 до 366,85; p=0,009). Низкая распространенность лейденской мутации в изученной группе (как и в целом в российской популяции) не позволила выявить достоверных различий.
Независимая связь между гомозиготным носительством полиморфного маркера C-426T гена фактора V и наличием тромба в ушке ЛП обнаружена впервые. Ранее мы выявили связь гомозиготного носительства полиморфного маркера C-426T с атеротромбозом [79]. Изученный маркер расположен в промоторной области и, следовательно, может влиять на уровень экспрессии гена. Возможно, изменение именно скорости синтеза кодируемого белка, а не его структуры, может играть значимую роль в патогенезе тромбообразования в ушке ЛП у больных с МА. По-видимому, сегодня уже достаточно данных для того, чтобы обсуждать воздействие на фактор V как одну из возможных целей антитромботической терапии.
Риск развития ТЭО
В исследовании SPAF III, включившем 1531 больного, показано отсутствие связи лейденской мутации с риском развития ТЭО при МА [72]. В крупном проспективном регистре (n=13 559) также не удалось выявить связь между наличием лейденской мутации и риском развития инсульта у больных с МА [74].
В исследовании случай—контроль (336 больных с МА и 336 здоровых добровольцев) выявлена независимая связь полиморфного маркера гена протромбина G20210A с наличием МА, но на риск развития ТЭО носительство этого полиморфизма не оказывало достоверного влияния [75].
В работе E. Berge и соавт. изучена связь полиморфных маркеров генов факторов V, протромбина и метилентетрагидрофолатредуктазы с риском развития ТЭО при МА [73]. В исследование включены 367 больных с ишемическим инсультом на фоне МА и 482 здоровых донора. В работе не выявлено достоверной связи изученных маркеров с риском развития инсульта. Среди носителей лейденской мутации отмечалось статистически недостоверное повышение риска раннего рецидива инсульта.
Выявлена связь полиморфного маркера A6534G гена β-цепи фибриногена с риском смерти от инсульта у больных с МА. Данный полиморфный маркер приводит к замене Тре на Ала в позиции 312 белка β-цепи фибриногена и влияет на способность фактора ХIIIа связываться с фибрин-мономерами. Фактор XIIIa, катализируя полимеризацию фибрин-мономеров, повышает устойчивость фибрина к протеолитической деградации плазмином. Теоретически данный полиморфизм может влиять на стабильность тромба. А. Carter и соавт. [76] изучали связь полиморфного маркера A6534G с риском развития ишемического инсульта, типами инсульта и смертностью от инсульта (519 больных инсультом и 423 здоровых донора). Не выявлено достоверной связи данного полиморфизма с риском развития инсульта и типами инсульта как у больных с МА (n=110), так и у больных с синусовым ритмом. Наличие аллеля G достоверно ухудшало прогноз у больных с МА. Выживаемость носителей генотипа А/А составила 42,1%, носителей генотипа A/G — 18%, а носителей генотипа G/G — 0 в течение 3-летнего наблюдения. Предполагается, что высокая смертность носителей аллеля G обусловлена снижением стабильности тромба и склонностью к эмболиям. Связь полиморфного маркера A6534G гена β-цепи фибриногена с наличием тромба в ушке ЛП не изучалась.
Другой часто изучаемый полиморфизм — полиморфный маркер G(-455)A гена β-цепи фибриногена, расположенный в промоторной области. Наличие аллеля —455A определяет более высокий уровень транскрипции гена. В работе V. Bozdemir и соавт. [80] обследованы 47 больных с МА, у 24 из них выявлен тромб в ушке ЛП. Распространенность аллеля —455A в группе больных с тромбозом ушка ЛП составила 37,5%, в группе без тромба — 15,1% (различия статистически незначимы). При добавлении к первой группе больных с выраженным спонтанным контрастированием различия достигали статистической значимости — 44,4% по сравнению с 10% (р=0,01). В нашей работе обследованы 53 больных с МА, у 11 из них выявлен тромбоз ушка ЛП. Нами не выявлено влияния этого маркера на развитие тромбоза при МА [73].
Заключение
Итак, тромб в левом предсердии при мерцательной аритмии образуется в связи с изменением гемодинамики, которое приводит к постепенному преобладанию процессов свертывания крови над противоположными реакциями. Большое значение имеет состояние эндокарда, при этом локальные воспалительные реакции могут существенно усугублять ситуацию. Генетический полиморфизм некоторых факторов системы гемостаза может оказать значимое влияние именно в случае, если равновесие в этой системе подвергается значимому давлению внешних обстоятельств. Расшифровка механизмов тромбообразования при мерцательной аритмии в будущем даст информацию для поиска новых направлений в целях медикаментозного предотвращения риска развития тромбоэмболических осложнений.



{kind=link}
{kind=link}