
Сосудистые заболевания по-прежнему являются ведущей причиной заболеваемости и смертности в мире, основной причиной инфаркта миокарда и ишемии [1]. Природа сосудистых заболеваний сложна и до сих пор недостаточно изучена. И хотя ремоделирование артерий (РА) можно рассматривать как естественный механизм старения сосудов, неблагоприятное ремоделирование, суживающее просвет артерии, ассоциируется со значительными гемодинамическими изменениями, сердечно-сосудистой заболеваемостью и смертностью [2]. РА осуществляется рядом сложных механизмов, которые тесно взаимосвязаны и воздействуют как на клеточные, так и неклеточные компоненты сосудистой стенки. Механизмы, участвующие в ремоделировании сосудистой стенки, включают гиперплазию интимы и медии, изменения внеклеточного коллагена, эластина, функции эндотелия и фиброз [3]. Миграция и пролиферация сосудистых гладких мышечных клеток (ГМК) способствуют утолщению интимы и медии артерий. Дифференцировка ГМК из сократительного в секреторный фенотип приводит к повышению тонуса сосудов и способствует накоплению внеклеточного матрикса (ВКМ). Из-за сложности и множественности процессов трудно обнаружить единый механизм, способствующий неблагоприятному ремоделированию сосудов. Многолетние исследования функций активатора плазминогена урокиназного типа (урокиназы), а также его влияния на различные патофизиологические звенья РА привели нас к заключению, что именно урокиназа является ключевым регулятором ремоделирования в стенке сосудов после механического повреждения [4].
РА: общие аспекты. РА является перестройкой сосудистой стенки, которая отражает адаптацию сосуда к механическим и гемодинамическим стимулам [5]. РА сопровождается изменениями в структуре и функции сосудистой стенки и наблюдается при атеросклерозе, а также заживлении сосудов после повреждения. Атеросклероз характеризуется фокальным воспалительным процессом в интиме и накоплением липидов в атеросклеротических бляшках (АСБ), а артериосклероз — диффузным поражением сосудистой стенки [6].
Артериосклероз связан со старением, сердечно-сосудистыми, метаболическими или воспалительными заболеваниями [5]. Макроскопически можно выделить различные типы перестройки сосудистой стенки в зависимости от типа и локализации сосуда (рис. 1, см. цветную вклейку). РА может сопровождаться как расширением (положительное ремоделирование), так и сужением (отрицательное ремоделирование) просвета артерии, может быть гипертрофическим (утолщение стенки сосуда), эутрофическим (нормальная толщины стенки) или гипотрофическим (истончение сосудистой стенки) [7]. Типичные для артериосклероза изменения наблюдаются в основном при перестройке крупных центральных артерий эластического типа. Они характеризуются увеличением диаметра сосудов и утолщением интимы и медии (положительное гипертрофическое ремоделирование) [8]. В то же время ремоделирование периферических сосудов мышечного типа чаще бывает отрицательным эу- или гипертрофическим, что, вероятно, отражает устойчивые вазоконстрикторные влияния [9]. В случае заживления сосуда после внутрисосудистого повреждения (например, после ангиопластики, стентирования) ремоделирование может быть как эу-, так и гипертрофическим, в случаях дезадаптивного ответа является отрицательным, т.е. приводит к сужению просвета артерии, как это происходит при развитии рестеноза.
Адаптивное ремоделирование сосудов (также известное как положительное ремоделирование, или феномен Глагова) впервые было обнаружено в коронарных артериях (КА) на ранних стадиях атеросклероза, где компенсационное расширение сосудов имеет место для поддержания постоянного кровотока, несмотря на рост АСБ [11].
Эта адаптация зависит от динамического взаимодействия между факторами роста, вазоактивными веществами и гемодинамическими стимулами (потоком, растяжением, напряжением сдвига), приводит в конечном итоге к устойчивым изменениям просвета или относительного состава сосудистой стенки (положительное ремоделирование) [12]. Если эта реакция выходит за рамки возможностей адаптации, ремоделирование становится патологическим и приводит к уменьшению размеров просвета артерии (отрицательное, или конструктивное ремоделирование). Это вносит вклад в развитие сердечно-сосудистых заболеваний (атеросклероз, рестеноз и недостаточность шунтов) и в конечном итоге может привести к клиническим осложнениям, таким как ишемия миокарда или нижних конечностей, или инсульт [13].
За положительное ремоделирование, главным образом, отвечает активность протеаз (в основном матриксных металлопротеиназ — ММП) в АСБ, стимулированная воспалением. Этот феномен вызывает потерю коллагена и уменьшение количества ГМК [12], что приводит к истончению медии и адвентиции и тем самым обусловливает внешнюю экспансию сосудистой стенки [14]. Аналогичные, но более интенсивные процессы происходят при формировании аневризм сосудов [15]. Следует заметить, что ММП также способствуют нестабильности АСБ [16]. Этот тип ремоделирования таит в себе парадоксальную опасность: хотя он задерживает сужение просвета, это может повысить риск разрыва АСБ и тромбоза с последующим острым сужением просвета или даже окклюзией артерии [12].
Отрицательное ремоделирование является развивающимся во времени феноменом, в значительной степени напоминающим ранозаживление. Утолщение стенки артерии вызывают гиперплазия ГМК интимы и медии, а также гиперплазия клеток адвентиции и отложение ВКМ в слоях сосудистой стенки [17]. При РА меняются нормальный состав и укладка ВКМ в сосудистой стенке. В медии нормальной артериальной стенки эластические волокна расположены параллельными, концентрическими, фенестрированными слоями, чередующимися со слоями ГМК, заякоренными к эластическим волокнам гликопротеинами и интегринами [18]. Эти структуры, называемые «эластические мембраны», позволяют сосуду расширяться в ответ на пульсовую волну систолического артериального давления и обеспечивают пассивный упругий буфер, в то время как ГМК динамически перераспределяют напряжение растяжения волокон благодаря способности сокращаться и расслабляться [19]. При РА слоистая архитектура эластических мембран теряется, и они постепенно становятся фрагментарными и фиброзными [20]. С возрастом ГМК синтезируют в основном не эластин, а неэластичный коллаген, увеличивающий жесткость сосудистой стенки [21]. Отложение минералов кальция дополнительно способствует жесткости и неблагоприятному ремоделированию сосудистой ткани [22]. При механическом повреждении сосуда под действием различных факторов может происходить усиленный синтез коллагена клетками сосудистой стенки как ГМК, так и миофибробластами, что ведет к локальному утолщению сосудистой стенки и сужению просвета артерии, т.е. отрицательному ремоделированию в участке повреждения [23].
Перестройка сосудистой стенки в ответ на различные воздействия включает два вида основных процессов: изменение структуры и толщины интимы—медии (ТИМ) и геометрическое РА. От соотношения этих процессов зависит конечный физиологически и клинически важный результат — размер просвета артерии. Один из наиболее важных показателей сосудистого ремоделирования — утолщение интимы—медии сонных артерий (СА), регистрируемое при ультразвуковом исследовании, является фактором риска развития сердечно-сосудистых заболеваний (ССЗ) и их осложнений [24]. Утолщение интимы—медии играет компенсаторную роль для обеспечения адекватности кровотока и коррелирует с размером просвета артерии [25]. Важным является то, что увеличение индекса интима—медиа общей СА до 1,2 мм сопровождается пропорциональным увеличением просвета сосуда, дальнейший рост индекса (>1,3 мм) ведет к обратному процессу — концентрическому сужению просвета артерии. Ультразвуковые признаки утолщения стенки артерий вошли в Европейские рекомендации по профилактике, диагностике и лечению артериальной гипертонии (АГ) как одна из характеристик поражения органов-мишеней [26]. Тесная взаимосвязь утолщения стенки СА и риска развития кардиальных и цереброваскулярных осложнений ассоциируется с высокой распространенностью повышенных значений ТИМ у пациентов с высоким риском развития сердечно-сосудистых осложнений в отсутствие симптомов [27]. Высокая прогностическая значимость утолщения интимы—медии определяет важную роль сосудистого ремоделирования в развитии и прогрессировании ССЗ.
Патогенез РА. РА обусловлено множеством взаимосвязанных и сложно регулируемых процессов. Процессы, которые имеют особое значение, поскольку являются ключевыми, включают пролиферацию и дифференцировку клеток, деградацию волокон эластина и отложение ВКМ, нарушение функции эндотелия, важность которой трудно переоценить.
Адвентиция артерии считалась поддерживающей тканью по сравнению с интимой и медией, однако экспериментальные данные последних лет показали, что повреждение адвентиции индуцирует патологические изменения в интиме и медии, подобные тем, которые вызывает повреждение эндотелия. Кроме того, было показано, что изменения в адвентиции участвуют в развитии ремоделирования интимы и медии [28]. Миофибробласты имеют решающее значение для заживления и развития фиброза сосудистой стенки и играют важную роль в ремоделировании адвентиции [29].
Множество патологических состояний, например АГ [7], а также различные виды сосудистых повреждений: либо внутрипросветное (например, формирование АСБ [11], перерастяжение сосуда при баллонной дилатации) [30], или периваскулярное (например, периваскулярное сдавление) [31] могут нарушать структуру и функцию сосудистой стенки. В ответ на эти стимулы включаются относительно стандартные механизмы адаптации и заживления, ведущие к ремоделированию адвентиции .
В поврежденных баллонным катетером КА было показано, что отрицательное ремоделирование, в том числе опосредованное усиленным делением фибробластов адвентиции и трансформацией фибробластов в миофибробласты, приводит к образованию утолщенной адвентиции, богатой миофибробластами и коллагеновыми волоконами. В конце концов, адвентициальные миофибробласты подвергаются быстрому апоптозу, образуя рубцовоподобную ткань, сдавливающую сосуд [13, 29]. Помимо сдавления реорганизация коллагеновых волокон, межмолекулярные сшивки, а также непрерывный синтез и отложение эластина играют роль в ремоделировании слоев сосудистой стенки и ведут к сужению просвета КА [32]. Отрицательное ремоделирование в связи с модификацией адвентиции является одним из основных факторов, определяющих развитие рестеноза после баллонной ангиопластики, помимо раннего эластического спазма и роста неоинтимы [33–36]. Стенты устраняют негативное ремоделирование, но вместе с тем приводят к чрезмерному росту интимы [33].
Функция ГМК имеет первостепенное значение для понимания причин РА. В нормальных артериях ГМК медии регулируют диаметр и тонус сосудов для поддержания гемодинамического равновесия [37].
Для выполнения этой регуляторной функции ГМК должны иметь сократительной фенотип. ГМК сократительного фенотипа характеризуются рядом специфических маркерных белков, таких как гладкомышечный 22α (SM22α), α-актин гладких мышц (αSMa) и смузелин [38]. Определенное количество ГМК медии способны дифференцироваться в клетки синтетического фенотипа, которые могут затем делиться и мигрировать или секретировать ВКМ [39]. Фенотипическая гибкость ГМК необходима для адаптации сосудистой стенки к различным условиям. Различные стрессовые сигналы переключают экспрессию генов, модулирующих фенотип ГМК для адаптации. Этот процесс дифференцировки называется фенотипическим переключением и считается важным механизмом РА [37, 38]. Фенотипическое переключение происходит в ответ на повреждение сосудов или стресс и характеризуется снижением экспрессии генов специфических для сократительного фенотипа ГМК [37].
ГМК синтетического фенотипа продуцируют протеазы, расщепляющие эластин (в том числе активаторы плазминогена, ММП). Эластические волокна состоят из полимеров тропоэластина, сшитых с микрофибриллами, богатыми фибриллином. В сосудистой стенке эластин в основном синтезируется внутриутробно и в период новорожденности секреторными ГМК. При старении происходит разрушение эластических волокон, ведущее к ремоделированию и жесткости сосудов [40]. Высказано предположение, что деградация эластина является преимущественно результатом усталости вследствие циклического растяжения эластических волокон с каждым ударом сердца [5, 8]. В поддержку этой гипотезы показано, что структурные изменения эластина пропорционально связаны с общим количеством циклов сердца [41]. Кроме того, синтез коллагена ГМК и миофибробластами обеспечивает стабильность АСБ. Напротив, если значительная часть ГМК имеет синтетический фенотип и секретирует протеолитические ферменты, деградация фиброзной покрышки может способствовать разрыву АСБ [42]. Таким образом, стабильность АСБ во многом зависит от экспрессии протеолитических ферментов [43].
Протеазы облегчают клеточную миграцию, отделяя клетки от базальной мембраны и ВКМ. Гиперэкспрессия ММП совпадает с миграцией ГМК [44]. M.P. Bendeck и соавт. показали, что ингибирование активности ММП подавляет миграцию сосудистых ГМК [45]. Промиграционные стимулы воздействуют на цитоскелет ГМК. Ламеллоподии вытягиваются из лидирующего края клетки за счет полимеризации актина, что позволяет ей перемещаться через ВКМ по направлению к хемотактическому стимулу [44]. Миграция способствует перемещению ГМК из медии в интиму и приводит к сужению просвета артерии. Особую роль в регуляции миграции клеток сосудистой стенки играет активатор плазминогена урокиназного типа (урокиназа) [3, 4].
Функция эндотелия играет важнейшую роль в РА. Функция эндотелия меняется с возрастом, а дисфункция эндотелия (ДЭ) сопровождает многие ССЗ. Приток крови и напряжение сдвига влияют на синтез эндотелиальными клетками оксида азота (NO), который регулирует сокращение и расслабление ГМК. В ответ на патологические воздействия, такие как изменения напряжения сдвига, воспаление или внутрисосудистое повреждение, эндотелиальные клетки экспрессируют цитокины и факторы роста, которые влияют на гомеостаз сосудистой стенки [34, 35]. Цитокины и факторы роста в свою очередь регулируют пролиферацию, миграцию и трансдифференцировку ГМК и фибробластов, а также синтез ВКМ этими клетками [2, 36]. Отложение белков ВКМ, таких как фибронектин и фибриноген, способствует пролиферации, выживанию клеток и ремоделированию сосудистой стенки.
При колебаниях артериального давления РА на первых этапах является компенсаторным и служит для снижения напряжения в стенке артерии, однако на более поздних стадиях компенсаторные механизмы становятся патологическими и инициируют патофизиологические отклонения. Так, фрагментация эластической мембраны, гиперплазия и гипертрофия ГМК, потеря сократительной способности ГМК, отложение коллагена и кальцификация ведут к жесткости артерий. Многие исследования показали, что жесткость артерий независимо связана с риском развития ССЗ и смерти от них, а также является независимым предиктором поражения органов-мишеней: сердца, почек и мозга [46, 47]. Жесткость артерий отражает степень ремоделирования крупных артерий и используется в качестве параметра для стратификации риска развития ССЗ наряду с традиционными факторами риска [48].
В нормальных условиях ламинарный поток крови и циклические напряжения сдвига поддерживают адекватные функции эндотелия, такие как NO-опосредованная регуляция сосудистого тонуса, предотвращение тромбоза и воспаления, поддержание нормального метаболизма ВКМ и регуляция проницаемости сосудов [49]. При ремоделировании турбулентный кровоток и локальные изменения напряжения сдвига вызывают ДЭ, которая характеризуется нарушением синтеза NO и активацией провоспалительных и проатерогенных факторов, усилением окислительного стресса, а также вазоконстрикцией [2, 49]. Жесткость артерий и ДЭ не только стимулируют развитие АСБ, но и способствуют дальнейшей реконструкции медии сосудов, а также приводят к повреждению органов-мишеней, таких как почки, мозг и сердце.
Сосудистые клетки могут чувствовать изменения механических сил и преобразовывать механический сигнал в биологический ответ — феномен механотрансдукции, механизмы которого неясны. Предложено несколько механизмов, обеспечивающих трансформацию механических стимулов, это стретч (растяжение)-активируемые ионные каналы, интегрины, цитоскелет, рецепторы тирозиновых киназ [50, 51], а также внутриклеточная сигнализация, запускаемая активными формами кислорода (АФК) [52]. Исследования показали, что изменения уровней напряжения сдвига или растяжения вызывают выраженные изменения в уровне АФК, которые могут способствовать спазму сосудов при АГ [52], вызывать дисфункцию сосудов малого круга кровообращения [53] или стимулировать РА, либо ангиогенез через АФК-опосредованную активацию транскрипции факторов роста, белков ВКМ, ММП или биоактивных пептидов [54, 55].
В эндотелиальных клетках АФК являются важными вторичными посредниками в регуляции роста клеток, миграции, пролиферации и выживания [56]. При нарушениях напряжения сдвига происходит усиление АФК-зависимой сигнализации за счет подавления антиоксидантных генов и гиперэкспрессии секретируемых факторов [57]. Изменения кровотока приводят к устойчивой гиперпродукции АФК, индукции оксидативного стресса и активации в эндотелиальных клетках MAP-киназ и NF-κB, что способствует ДЭ и воспалению [58]. Окислительный стресс также связан с образованием окисленных липопротеидов низкой плотности, которые могут стимулировать активацию многих провоспалительных сигнальных путей и усиливать атерогенез [59].
Таким образом, нормальные адаптивные ответы эндотелия на нарушения кровотока, опосредующие сосудистый гомеостаз, могут стать неадекватными в условиях, когда нарушения напряжения сдвига становятся хроническими, а также в присутствии других факторов риска [60].
Воспаление играет важную роль в РА, особенно при атеросклеротическом поражении. Клетки воспалительного типа привлекаются в участок повреждения и экспрессируют активные вещества, способствующие миграции и пролиферации клеток сосудистой стенки.
Привлечение моноцитов/макрофагов молекулами клеточной адгезии, такими как ICAM-1 и VCAM, чувствительно к изменениям напряжения сдвига [61], что отчасти объясняет преобладание макрофагов и Т-клеток выше пораженных атеросклерозом участков [62] и в сосудах, в которых преобладает позитивное ремоделирование [63]. Гиперлипидемия также увеличивает инфильтрацию воспалительными клетками АСБ и способствует экспрессии этими клетками ММП. Бóльшая часть экспрессии ММП в АСБ обеспечивается макрофагами/пенистыми клетками и может быть уменьшена за счет снижения уровня липидов или подавления окисления липидов [64]. С повышенной локальной активностью ММП связывают положительное ремоделирование и истончение медии, наблюдаемые при атеросклерозе [65]. При повреждении АСБ, например после ангиопластики или разрыве АСБ, образование фиброзного рубца может привести к негативному ремоделированию и сужению просвета артерии [66].
Ремоделирование сосудов при атеросклерозе и рестенозе. Атеросклеротическое поражение сосудов проявляется увеличением ТИМ и прямо коррелирует с уровнем холестерина липидов промежуточной и низкой плотности [67]. Данные экспериментальных исследований свидетельствуют, что при атеросклерозе первично возникает утолщение интимы. Установлена также связь увеличения ТИМ с нарушениями кальциевого и фосфорного метаболизма, активностью паратгормона и возрастом [68]. Благодаря большому просвету проводящих артерий базальный кровоток в них не изменяется до тех пор, пока сужение диаметра не достигнет 50%. Он снижается лишь при сужении просвета сосуда более чем на 70—80%, что означает критический стеноз. При этом снижается способность к увеличению кровотока при нагрузке, в связи с чем ишемия миокарда, развитие церебральных катастроф и поражение периферических артерий являются частыми осложнениями у таких больных [69].
В 1964 г. C.T. Dotter и M.P. Judkins предложили принципиально новое решение хирургического лечения стенозирующих осложнений при атеросклерозе [70]. Авторы выполнили механическое внутрисосудистое расширение бедренной артерии при помощи коаксиального катетера. В дальнейшем G. Porstmann провел ряд экспериментов с использованием латексного баллона и внутрисосудистого катетера [71]. В 1977 г. в Цюрихе A.R. Gruentzig выполнил первое вмешательство по этой методике, применив внутрисосудистую баллонную ангиопластику в клинике [72].
Проведение транслюминальной баллонной коронарной ангиопластики (ТБКА), действительно, уменьшало стеноз КА, а также устраняло объективные и субъективные проявления ишемической болезни сердца (ИБС) [73]. Раздувание баллона сопровождалось фокальным разрушением АСБ и ее компрессией с распределением компонентов АСБ по стенке сосуда. Вокруг атеромы возникали надрывы интимы и медии, которые проявлялись при коронарографии более или менее выраженной диссекцией тканей. Закрытие дефекта тканей КА новым эндотелием с восстановлением целостности стенки артерии происходило через несколько недель после вмешательства [74]. После успешного проведения ТБКА появилось значительное число больных, у которых отмечалось возобновление стенокардии спустя 4—6 мес. Было установлено, что чрезмерная выраженность процесса восстановления целостности стенки КА приводит к ее повторному сужению — рестенозу [75, 76]. Повторное сужение артерии — патология, которая возникает после вмешательства и до сих пор считается ведущей проблемой эндоваскулярной хирургии.
Гистологически рестеноз на месте выполненной ангиопластики определяется выраженным разрастанием неоинтимы и гемодинамически значимым сужением просвета артерии. Наиболее распространенным показателем ангиографического рестеноза служит сужение просвета КА более чем на 50%, которое определяется при повторной ангиографии после успешно проведенной ТБКА через 3—6 мес [76]. В среднем частота рестенозов после ТБКА составляла, по разным данным, от 10 до 40% [77]. Рестеноз после ТБКА, являющейся одной из основных техник реваскуляризации, является результатом сложного процесса заживления сосудистой стенки после повреждения, также включающего каскад клеточных и молекулярных событий и высвобождение множества вазоактивных, тромбогенных и митогенных факторов [78]. Сразу после ангиопластики в 30% случаев происходит острый спазм эластических компонентов сосудистой стенки, который приводит к раннему сужению просвета артерии.
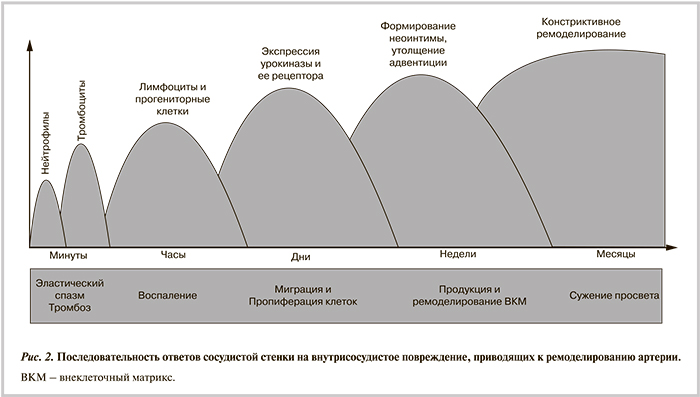
В то же время рестеноз служит причиной позднего сужения просвета КА в течение 6—9 мес и происходит за счет интенсивного пролиферативного и миграционного ответов сосудистой стенки на локальное повреждение, а также сдавления сосуда за счет негативного ремоделирования [78, 79]. В соответствии с существующими представлениями, рестеноз представляет собой недостаточно адаптированный ответ артерии на травму, вызванную ангиопластикой, которая запускает серию процессов, таких как тромбоз, воспаление, миграция и пролиферация клеток и избыточная продукция ВКМ (рис. 2). Все это вместе приводит к сужению просвета КА после процедуры [78, 79]. Помимо этого на участке дилатации возможно тромбообразование [80].
При расширении просвета сосуда баллонным катетером повреждаются клетки эндотелия, возможны радиальные разрывы гладкомышечных слоев и продольные эластических мембран интимы и медии [78, 81]. Особенно легко подвержены разрывам области АСБ [81]. В результате повреждения сосудистой стенки проницаемость сосудов повышается, активируется каскад системы свертывания [82]. Обнажение субэндотелиального матрикса вызывает адгезию тромбоцитов и образование окклюзирующего тромба. Этот процесс может продолжаться в течение 48 ч после ангиопластики [83]. Пристеночный тромб на участке повреждения сосуда образуется сразу после вмешательства и может способствовать развитию рестеноза в отдаленном периоде. Дегрануляция тромбоцитов сопровождается высвобождением прокоагулянтных, вазоконстрикторных и митогенных факторов, включая тромбин, тромбоцитарный фактор роста (PDGF), тромбоксан А2, серотонин, фактор Виллебранда, аденозиндифосфат, фибронектин, фактор V. PDGF — мощный стимулятор пролиферации и миграции ГМК [84]. Тромбин и другие белки системы свертывания крови также являются митогенами и хемоаттрактантами для ГМК и моноцитов [85]. Например, ингибирование тромбина приводит к уменьшению размеров неоинтимы после экспериментальной ангиопластики [86].
У человека наличие ангиографических признаков тромбоза после ангиопластики коррелирует с повышенной вероятностью развития рестеноза [87], однако использование антикоагулянтов и антиагрегантов для профилактики рестенозов пока нуждается в дальнейших исследованиях [88]. Образование сгустка из фибрина и тромбоцитов способствует адгезии и миграции лейкоцитов из крови в поврежденную стенку артерии. В привлечении лейкоцитов участвует Р-селектин, экспонируемый на поверхности активированных тромбоцитов [89]. В свою очередь лейкоцитарные b2-интегрины обеспечивают прочное прикрепление моноцитов и гранулоцитов к фибрину и тромбоцитарным гликопротеинам [90], b1-интегрины опосредуют адгезию моноцитов на фибронектине [91]. В моделях рестеноза у животных реактивные воспалительные инфильтраты обнаруживаются по всей толщине артериальной стенки [92]. Нейтрофильные гранулоциты обеспечивают неспецифический ответ стенки сосуда на травму в самые ранние сроки, а привлеченные моноциты, трансформирующиеся в макрофаги, присутствуют в сосудистой стенке в течение нескольких недель [93].
В участке повреждения артерии из активированных тромбоцитов, лейкоцитов и ГМК происходит мощный выброс митогенов и белков, способствующих миграции и пролиферации клеток [94]. Волна пролиферации ГМК медии, называемая первой волной [78], обусловлена выделением основного фактора роста фибробластов (bFGF) из погибающих клеток [95].
Гиперплазия неоинтимы является в значительной степени результатом миграции активированных ГМК медии к участку повреждения и синтеза этими клетками ВКМ [96]. В регуляции этого процесса участвуют PDGF, трансформирующий β-фактор роста (TGF-β), bFGF и ангиотензин II [97], которые продуцируются тромбоцитами и моноцитами. Гиперплазия интимы, как правило, ассоциирована с транформацией активно делящихся ГМК из сократительного в синтетический фенотип [98]. Эта фенотипическая трансформация ассоциируется с повышенной продукцией компонентов ВКМ, таких как протеогликаны и коллаген, которые обладают способностью регулировать пролиферацию и дифференцировку клеток [99]. В некоторых исследованиях получены данные об участии стволовых клеток кост-ного мозга в развитии неоинтимы после повреждения [100, 101]. Предполагается, что стимул к пролиферации клеток неоинтимы опосредован участием PDGF [102], рецептора ангиотензина II АТ-1 [103], TGF-β [104]. Тем не менее к настоящему моменту в клинической практике не доказана возможность блокады пролиферации с помощью антагонистов какого-либо из перечисленных факторов.
По мере регенерации эндотелия пролиферация клеток неоинтимы замедляется, а быстрая реэндотелизация ограничивает развитие неоинтимы [105]. В сосудах с выраженной неоинтимой часто отмечают неполную реэндотелизацию [106]. В моделях на животных попытки ускорить реэндотелизацию и таким образом ограничить рост неоинтимы с помощью трансфекции гена фактора роста гепатоцитов, избирательно стимулирующего рост эндотелия, но не ГМК, имплантации эндотелиальных клеток или внутривенного введения предшественников эндотелиальных клеток оказались весьма успешными [107]. В качестве основного фактора, тормозящего пролиферацию ГМК, рассматривается синтезируемый эндотелиальными клетками NO [108].
Отрицательное РА является одним из основных механизмов позднего сужения просвета артерии после ангиопластики без стентирования [109, 110]. На животных моделях баллонной ангиопластики показано, что компенсаторное расширение сосуда является важной детерминантой размеров просвета после баллонирования [111]. Результаты многих исследований указывают на то, что вазоконстрикция также может быть одним из механизмов рестенозирования [109, 112, 113]. M.J. Post и соавт., используя 3 различные животные модели и 2 разных типа внутрисосудистого повреждения, обнаружили, что ремоделирование в большей степени, чем гиперплазия неоинтимы, определяет развитие рестеноза [112]. Обнаружена значимая связь между ответом адвентиции и хронической констрикцией артерии после экспериментального баллонирования [113]. Ответ на вызванное растяжением повреждение адвентиции приводит к фиброзу адвентиции [114, 115] и сдавлению сосуда вокруг зоны повреждения [109, 115]. Показано, что глубокое повреждение сосуда с разрывами адвентиции приводит к утолщению адвентиции, трансформации фибробластов в миофибробласты, их пролиферации и активной выработке внеклеточного коллагена [115]. Такой ответ адвентиции может играть роль в развитии рестеноза после баллонной ангиопластики за счет кольцевого сдавления сосуда или предупреждения расширения наружной эластической мембраны [109]. В КА пациентов с выраженным атеросклеротическим поражением выявлена корреляция между толщиной адвентиции и исходом ТБКА, по данным гистологических исследований [116].
Необходимо остановиться на процессах перестройки сосудистой стенки, приводящих к развитию рестеноза, после ангиопласти с имплантацией стента. Для увеличения эффективности ТБКА была разработана новая техника эндоваскулярной реваскуляризации с установкой внутрисосудистого стента. Тем не менее риск формирования рестеноза стента остается достаточно высоким — от 10 до 40%, по данным разных авторов [117], что лимитирует эффективность стентирования в отдаленные сроки. По данным проведенных к настоящему времени многоцентровых исследований [118, 119], независимыми предикторами рестеноза принято считать наличие сахарного диабета, протяженные поражения (более 20 мм), малый диаметр сосуда (менее 2,5 мм).
Основной механизм рестеноза после баллонной дилатации — ремоделирование стенки сосуда, тогда как рестеноз после имплантации эндопротеза — гиперпластическая реакция гладкомышечных элементов стенки сосуда в ответ на травму и инородное тело [120] (рис. 3). Формирование рестеноза стента происходит в более поздние сроки по сравнению с баллонной ангиопластикой — через 6—8 мес после процедуры, а не через 3—6 мес. По данным гистологических исследований, пролиферация ГМК различной степени выраженности в месте рестеноза стента наблюдается в 40—50% случаев. Помимо этого, как правило, отмечают миграцию и скопление в месте имплантации стента клеток лейкоцитарного ряда: макрофагов, нейтрофилов, В-лимфоцитов [121]. При гистологических исследованиях морфологического субстрата рестеноза во всех случаях наблюдается увеличение ВКМ с накоплением в нем протеогликанов [122]. Таким образом, формирование рестеноза в месте имплантации стента происходит за счет пролиферации клеток, воспалительной инфильтрации и синтеза ВКМ.
Наиболее перспективным подходом к лечению рестеноза в настоящее время считается создание технологии локальной доставки лекарственного вещества с помощью внутрисосудистого эндопротеза. Опыт клинического применения имеют следующие типы лекарственных препаратов для покрытия коронарных эндопротезов: противосвертывающие — гирудин, гепарин, абсциксимаб; ингибиторы миграции — ингибиторы С-протеиназы, ингибиторы металлопротеазы (Batimastat); ускоряющие заживление — ингибиторы ГМГ-КоА-редуктазы, 17-β-эстрадиол; противоопухолевые — паклитаксель, актиномицин D; иммунносупрессанты — рапамицин (сиролимус), дексаметазон [123]. Наибольший опыт клинического применения имеют эндопротезы, покрытые препаратом рапамицин (сиролимус) из группы иммунносупрессантов. Рапамицин представляет собой макроциклический лактон, обладающий высокой антипролиферативной активностью. В исследовании SIRIUS после имплантации стентов, покрытых рапамицином, частота рестеноза составила 3,2% [124].
Накопленные к настоящему моменту данные о применении в клинической практике стентов с лекарственным покрытием свидетельствуют о высокой эффективности данного вида эндопротезов при вмешательствах на первичных поражениях. Дальнейшие разработки в этом направлении будут способствовать решению проблемы рестеноза, в частности рестеноза стента.
РА после внутрисосудистого повреждения как терапевтическая мишень. Патофизиологические звенья ремоделирования, такие как пролиферация и миграция ГМК, фенотипическая трансформация клеток, отложение ВКМ, кальцификация, воспаление, а также молекулы, регулирующие эти процессы, могут быть потенциальными кандидатами для вмешательства. Тем не менее найти оптимальную мишень для воздействий оказалось непростой задачей в связи с множеством регуляторных механизмов и молекул-участников. В наших исследованиях на модели ремоделирования поврежденной артерии нам удалось обнаружить уникальный регулятор большинства процессов, вовлеченных в ремоделирование сосудов при их повреждении. Это активатор плазминогена урокиназного типа (урокиназа).
К настоящему времени имеется много фактов, свидетельствующих о значении протеаз как факторов риска развития ИБС и других сосудистых заболеваний [82, 125]. Нарушения функционирования компонентов системы фибринолиза способствуют прогрессированию атеросклероза, так как формирование тромба на поврежденной сосудистой стенке ускоряет прогрессирование атеросклероза в неповрежденных артериях и способствует развитию рестеноза на участке повреждения [126]. В то же время повышенное содержание урокиназы в периферической крови больных, подвергающихся ангиопластике и стентированию КА, соотносится с высоким риском развития рестенозов и является предиктором ангиографически подтвержденного коронарного рестеноза [127]. Мы обнаружили, что антиген урокиназы и ее активность в плазме пациентов существенно выше у пациентов со стенокардией, чем у здоровых добровольцев [128], а также у пациентов с возвратом стенокардии и рестенозом после баллонной ангиопластики, чем без рестеноза [129]. Экспрессия и активность урокиназы повышены в ГМК и макрофагах атеросклеротических, а также рестенотических АСБ в артериях человека [126, 130, 131]. Так, содержание рецептора урокиназы на макрофагах и ГМК неоинтимы в АСБ в 9 раз выше, чем в нормальном сосуде [126]. Выявлено, что в АСБ урокиназу экспрессируют преимущественно активированные макрофаги [132]. В то же время известно, что «нестабильные», склонные к разрыву, потенциально наиболее опасные АСБ инфильтрированы пенистыми клетками и моноцитами/макрофагами [133]. Исследования in vivo показали, что повышение экспрессии урокиназы макрофагами ускоряет прогрессирование атеросклероза и раннюю смерть трансгенных мышей [134]. Помимо этого, являясь фактором хемотаксиса для ГМК, лейкоцитов и моноцитов/макрофагов урокиназа, экспрессированная в АСБ, может способствовать миграции ГМК, еще большему привлечению моноцитов/макрофагов в АСБ и, таким образом, приводить к росту и «дестабилизации» АСБ [135, 136].
Урокиназа может активировать ММП и высвобождать связанные с матриксом факторы роста, в частности TGF-β1, основной фактор роста фибробластов и фактор роста колоний гранулоцитов и макрофагов, которые активно участвуют в процессах атерогенеза [137].
В то же время сами факторы роста стимулируют миграцию и хемотаксис клеток и способны повышать экспрессию урокиназы моноцитами/макрофагами и ГМК [138].
Наши работы показали, что урокиназа является полифункциональным белком, регулирующим миграцию и пролиферацию клеток на разных уровнях и участвующим в формировании неоинтимы и ремоделировании сосудов при повреждении [139—144]. Несмотря на то что и тканевый активатор плазминогена (ТАП), и урокиназа активируют плазминоген с образованием плазмина, их роль в РА различна. После баллонного повреждения артерии экспрессия и ТАП и урокиназного активатора плазминогена ГМК медии быстро возрастает [145]. Обнаружены корреляции между ранней экспрессией урокиназы и пролиферацией ГМК, экспрессией ТАП и миграцией ГМК в поврежденных баллонным катетером артериях [145]. Мы показали, что экспрессия урокиназы повышается уже через 6 ч после повреждения и остается повышенной в течение 4 дней в медии и развивающейся неоинтиме [141], т.е. в течение времени, на которое приходится активная пролиферация ГМК в медии и их миграция в формирующуюся неоинтиму [145]. На модели РА, индуцированного снижением кровотока, мы также обнаружили, что содержание урокиназы коррелировало с ростом неоинтимы, тогда как содержание ТАП — с позитивным РА [146]. Мы также обнаружили, что синтез урокиназы клетками сосудистой стенки локально увеличен в АСБ [147]. В исследованиях на трансгенных животных [148] и артериях приматов [149] установлено, что урокиназа является ключевым фактором развития неоинтимы. Было выявлено, что отсутствие гена урокиназы, как и отсутствие генов обоих активаторов плазминогена, одновременно приводит к подавлению роста неоинтимы. В другом исследовании в отсутствие гена урокиназы ГМК неоинтимы были лишены способности мигрировать [150]. Неспособность ГМК дефицитных по урокиназе мышей к миграции, а следовательно, и к формированию неоинтимы, может быть обусловлена участием урокиназы в расщеплении ВКМ [148]. Трансгенная гиперэкспрессия урокиназы в интиме сосуда у кроликов с атеросклерозом стимулирует констриктивное РА [151]. На поверхности клетки урокиназа связывается с высокоафинным рецептором (uPAR/CD87), который локализуется на лидирующем крае мигрирующей клетки [152], а также с другими участками связывания [153]. Связывание урокиназы с рецептором uPAR/CD87 обеспечивает направленную локальную деградацию белков ВКМ по направлению движения клетки. Для установления изменений в структуре доменов урокиназы при ее взаимодействии с рецептором мы использовали метод ядерного магнитного резонанса. Полученные в работе данные впервые показали, что для специфического взаимодействия ростового домена с урокиназным рецептором достаточным условием является наличие компонента первичной структуры — Ω-петли в составе ростового домена, а не его вторичная β-структурная организация [154]. Эти результаты открывают новые возможности для направленного поиска антагонистов рецептора урокиназы путем конструирования пептидов, содержащих Ω-петлю ростового домена.
С использованием рекомбинантных форм урокиназы, которые были получены в Лаборатории генной инженерии РКНПК Минздрава РФ, а также нейтрализующих урокиназу антител, предоставленных Группой инженерной иммунологии РКНПК Минздрава РФ, мы обнаружили, что в отличие от нативной урокиназы протеолитически неактивная рекомбинантная урокиназа не стимулировала образование неоинтимы и неоадвентиции, вызывала менее выраженное, чем в контроле, утолщение медии и уменьшение просвета артерии. Ее эффект больше напоминал эффект нейтрализующих урокиназу антител, которые подавляли рост неоинтимы и предотвращали сужение просвета артерии, вызванное баллонированием [141]. Полученные данные позволили утверждать, что in vivo для стимуляции миграции клеток и РА преимущественное значение имеет протеолитическая активность урокиназы. Эти эффекты весьма специфичны для урокиназы и, вероятно, обусловлены не только ее способностью к образованию плазмина, так как ТАП оказывал противоположное действие [155]. Оценивая влияние экзогенной урокиназы на показатели геометрического РА после баллонирования, мы обнаружили, что она не только стимулирует рост неоинтимы, но вызывает констриктивное РА. ТАП, напротив, вызывал положительное ремоделирование наряду с подавлением роста неоинтимы [155]. Способность урокиназы вызывать констриктивное ремоделирование поврежденной артерии может быть обусловлена ее влиянием на адвентицию сосуда. Нанесение на адвентицию экзогенной урокиназы стимулирует ее рост, пролиферацию клеток, аккумуляцию α-актин-положительных клеток и моноцитарно-макрофагальную инфильтрацию, а нейтрализующие урокиназу антитела, напротив, уменьшают количество клеток сократительного фенотипа и подавляют рост неоадвентиции в ответ на повреждение [143]. При этом неактивная урокиназа (uPA-H/Q) и ТАП не оказывают стимулирующего влияния на аккумуляцию α-актин-положительных клеток и макрофагов в поврежденной адвентиции, что указывает на определяющее значение протеолитической активности урокиназы для осуществления этих процессов.
Во многих работах показана связь между РА и экспрессией ММП, осуществляющих деградацию ВКМ [44, 45]. Обнаружено, что развитие рестеноза через 6—8 мес после ТБКА у больных стабильной стенокардией ассоциировалось с повышением содержания ММП-2 и ММП-9 в плазме больных, а содержание ММП-9 у больных стабильной стенокардией коррелировало со степенью выраженности коронарного атеросклероза [156]. Ранее показано, что урокиназа может независимо от плазмина благодаря собственной протеолитической активности активировать ММП [157]. Мы показали влияние урокиназы на экспрессию ММП в клетках моноцитарных линий [158, 159] и фибробластов [160]. Кроме того, в нашем исследовании in vivo мы обнаружили, что активаторы плазминогена по-разному влияют на экспрессию и активацию ММП-2 и ММП-9 в баллонированной СА [140]. Увеличение в сосуде ТАП, напротив, уменьшает экспрессию ММП-2 и не влияет на образование активных форм этих ферментов. Этот факт позволил нам предположить, что урокиназа и ТАП могут по-разному влиять на экспрессию генов, кодирующих белки, которые участвуют в РА.
Для выяснения механизмов различного влияния активаторов плазминогена на ремоделирование сосудов мы провели исследование экспрессии генов в баллонированном сосуде при нанесении на него урокиназы и ТАП. Мы обнаружили, что урокиназа влияла на экспрессию обширной группы про-воспалительных генов и генов, регулирующих метаболизм митохондрий и оксидативный стресс [144]. Изменение экспрессии генов, участвующих в регуляции воспаления, под действием урокиназы и отсутствии подобных изменений под действием ТАП свидетельствует о том, что воспалительный каскад, вероятно, вовлечен в стимуляцию роста неоинтимы, констриктивное ремоделирование и как следствие — уменьшение просвета артерии под действием урокиназы.
Неспецифическая воспалительная реакция сосудистой стенки на повреждение — важное звено патогенеза как рестеноза, так и атеросклероза [161].
Вероятность развития рестеноза после ангиопластики у больных нестабильной стенокардией коррелирует с числом макрофагов в АСБ непосредственно перед вмешательством [162]. На модели экспериментальной баллонной ангиопластики мы показали, что привлечение в сосуд моноцитов может быть одним из механизмов влияния урокиназы на ремоделирование. Это подтверждается полученными нами данными о том, что урокиназа увеличивает экспрессию в стенке сосуда одного из основных провоспалительных факторов, секретирующихся моноцитами/макрофагами — фактора некроза опухолей α (TNF-α) и фермента, осуществляющего его превращение в активную форму (TACE — TNF-α converting enzyme) [163]. TNF-α является важным посредником развития воспаления и вызывает образование и секрецию провоспалительных факторов (интерлейкин-10, кортикостероиды, простагландины) [164].
Недавние исследования показали, что оксидативный стресс, возникающий в ответ на повреждение сосудистой стенки, играет важную роль в патогенезе ССЗ, поскольку способствует неблагоприятному ремоделированию сосудов [165]. Известно, что образование супероксид-радикала является одним из ключевых процессов, регулирующих сосудистый тонус и пролиферацию ГМК в стенке сосуда [166]. Повышенная экспрессия НАДФ-оксидаз в баллонированной артерии представляет собой важный механизм сигнализации, регулирующей рост неоинтимы, РА и атерогенез [167]. Мы обнаружили, что урокиназа стимулирует образование АФК и регулирует экспрессию генов, влияющих на оксидативный стресс, в поврежденной артерии после экспериментальной баллонной агиопластики in vivo [168]. Мы показали, что урокиназа способна повышать экспрессию НАД(Ф)H-оксидаз, продукцию АФК и регулировать пролиферацию ГМК как аутокринный фактор роста [169]. Недавно нами получены новые данные, расширяющие представления о сигнальных путях, регулируемых урокиназой. Мы показали, что урокиназа способна транслоцироваться в ядро и таким образом регулировать экспрессию генов [170]. Кроме того, недавно мы показали, что урокиназа может стимулировать ангиогенез в ишемизированных скелетных мышцах и миокарде [171]. Наши данные о стимуляции образования АФК и наличии редокс-зависимого механизма стимуляции пролиферации под действием урокиназы и данные о том, что урокиназа способна регулировать экспрессию генов, влияющих на оксидативный стресс в поврежденной артерии после экспериментальной баллонной агиопластики, выявляют новый механизм действия урокиназы, который может оказаться ключевым в регуляции роста неоинтимы и развитии неблагоприятного ремоделирования сосудистой стенки.
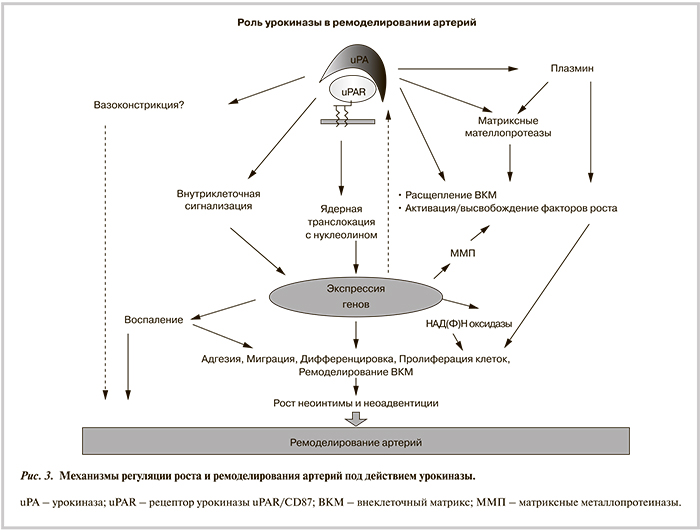
Многочисленные исследования показали, что урокиназа, являясь мультидоменным многофункциональным белком, регулирует адгезию, миграцию и пролиферацию клеток благодаря своей специфической протеолитической активности, образованию плазмина, расщеплению ВКМ, связыванию с рецепторами, взаимодействиям ее доменов друг с другом, с белками матрикса и интегринами, а также активации сложной внутриклеточной сигнализации. Многие эффекты урокиназы могут быть обусловлены ее ядерной транслокацией, взаимодействием с факторами транскрипции и ее влиянием на экспрессию генов. Все эти свойства урокиназы определяют ее ключевое значение в образовании и ремоделировании сосудов. Механизмы участия урокиназы в регуляции сосудистого роста и ремоделирования суммированы в следующей схеме (рис. 6).
С учетом роли урокиназы в сосудистом ремоделировании препараты, направленные на ингибирование урокиназы, при локальном использовании могут предотвращать рестенозирование после баллонной ангиопластики. Следует отметить, что антитела к урокиназе блокируют влияние на рост неоинтимы таких факторов роста, как PDGF, BFGF, HGF, VEGF. На основании этого мы обоснованно предположили, что урокиназа является ключевым звеном влияния многих факторов на процессы ремоделирования и регенерации. Недавно нами получены данные о подавлении спонтанной миграции эндотелиальных клеток и ангиогенеза на модели in vitro протеолитически неактивными рекомбинантными формами урокиназы uPA HQ и ее аминотерминальным фрагментом (АТФ) [172], что указывает на возможность использования созданных нами протеолитически неактивных рекомбинантных конструкций урокиназы для подавления неоангиогенеза в сетчатке глаза и опухолях. Полученные нами данные о ключевом значении первичной структуры «ростового» домена урокиназы для ее связывания с рецептором открывают новые возможности для создания антагонистов рецептора урокиназы и подавления эффектов эндогенной урокиназы в тканях, что может существенно улучшить перспективы контроля процессов роста и ремоделирования кровеносных сосудов. В сотрудничестве с Научно-исследовательским вычислительным центром МГУ им. М.В. Ломоносова, обладающим суперкомпьютером СКИФ МГУ «ЧЕБЫШЁВ» (пиковая производительность 60 трл. операций в секунду, объем оперативной памяти 5,5 Тб, 11-е место по производительности в мире) начаты поиск и разработка новых структур, подавляющих отдельные функции урокиназы.
Заключение
Ремоделирование сосудов представляет собой генерализованное или локальное изменение их размеров или структуры, которое не позволяет им только адаптироваться к изменяющимся условиям функционирования и восстанавливаться после повреждения, но и определяет патогенез и прогноз большинства сердечно-сосудистых заболеваний, включая атеросклероз, артериальную гипертонию, рестеноз, развивающийся после эндоваскулярной реваскуляризации, первичную легочную гипертензию, а также хронических заболеваний легких и почек [7]. Ремоделирование артерий является основным фактором, определяющим сужение просвета по всему сосудистому руслу. Клеточные и молекулярные механизмы патологического ремоделирования сосудистой стенки активно изучаются. Тем не менее механизмы, определяющие тип ответа и направление ремоделирования сосуда (положительное или отрицательное), остаются неясными. Ключевыми механизмами ремоделирования артерий являются миграция, пролиферация и апоптоз клеток сосудистой стенки, синтез и ремоделирование внеклеточного матрикса, а также воспаление и оксидативный стресс. Эти процессы управляются многочисленными биологически активными молекулами, включая цитокины, факторы роста и протеазы сосудистой стенки.
Последние два десятилетия ознаменовались бурным развитием и внедрением в клиническую практику лечения больных со стенозирующими заболеваниями сосудов нехирургических инвазивных вмешательств — ангиопластики и атерэктомии. Однако эффективность ангиопластик и атерэктомий значительно ограничивается быстрым развитием повторных стенозов (рестенозов). В основе развития стеноза/рестеноза лежит аномальная локальная пролиферация и миграция гладких мышечных клеток сосудистой стенки из медии в интиму, а также избыточный синтез белков внеклеточного матрикса этими клетками в ответ на повреждение сосуда. Эти процессы приводят к образованию неоинтимы и неоадвентиции, суживающих просвет сосуда.
Важным в отношении подходов к регуляции ремоделирования артерий является выбор специфической мишени для воздействий. В настоящее время проводятся разработки препаратов на основе множества активных молекул. Особый интерес вызывает урокиназа, так как она представляет собой многофункциональный мультидоменный белок, который не только регулирует фибринолиз, ремоделирование внеклеточного матрикса и миграцию и пролиферацию клеток, но также ассоциирован с развитием серьезных острых и хронических патологических состояний. Многие исследования как in vitro, так и in vivo указывают на особое значение урокиназы для негативного ремоделирования кровеносных сосудов, а также прогрессирования атеросклероза, нестабильности атеросклеротических бляшек и рестеноза [3, 4, 173]. Поскольку урокиназа является одним из важнейших участников реакции кровеносных сосудов на повреждение, это делает подавление ее эффектов перспективным подходом к лечению васкулопролиферативных процессов и прежде всего, рестенозов и окклюзий шунтов, развивающихся после эндоваскулярной и хирургической реваскуляризации. С этой целью может быть использовано блокирование взаимодействия урокиназы с клеточными рецепторами или внутриклеточных сигнальных путей, активируемых этим взаимодействием, или нейтрализация ее протеолитической активности. Результаты многолетних исследований свидетельствуют, что урокиназа является перспективной терапевтической мишенью.
Исследование выполнено за счет гранта Российского научного фонда (проект №14-24-00086)


