Гемобластозы представляют собой гетерогенную группу опухолевых заболеваний, морфологическим субстратом развития которых служат гемопоэтические клетки различной степени зрелости [1]. Верификация нозологических форм онкогематологических заболеваний основана на определении уровня дифференцировки кроветворных клеток и их принадлежности к клеточной линии. Согласно классификации ВОЗ 2016 г., в структуре миелопролиферативных заболеваний (МПЗ) выделяют группу так называемых Ph’-негативных (по филадельфийской хромосоме) болезней: хронический нейтрофильный лейкоз, истинная полицитемия, первичный миелофиброз (ПМФ), эссенциальная тромбоцитемия [2, 3]. В последние годы достигнуты значительные успехи в идентификации молекулярно-генетических механизмов развития МПЗ [3, 4]. Установлено, что наиболее важное диагностическое значение для подтверждения клонального процесса и оценки прогноза является выявление мутаций в генах JAK2, MPL, CALR, CSFR3R [3, 5]. При этом частота встречаемости данных мутаций значительно варьируется в группе Ph’-негативных заболеваний.
По данным литературы, частота встречаемости поражения почек при онкогематологических заболеваниях значительно варьируется [1, 5, 6]. Установлено, что при МПЗ почки в патологический процесс вовлекаются крайне редко [5], в то время как при лимфоплазмлоцитарных заболеваниях почки являются органом-мишенью [6].
Болезнь отложения легких цепей (БОЛЦ) и ПМФ относятся к редким гематологическим заболеваниям [3, 7]. Несмотря на различные патогенетические механизмы, лежащие в основе развития данных заболеваний, морфологические изменения, выявленные при гистологическом исследовании нефробиоптата, однотипны. Это подтверждает тот факт, что изображение нефробиоптата представляет собой морфологический профиль, а не диагноз.
Представляем клинические наблюдения, демонстрирующие особенности поражения почек при ПМФ и БОЛЦ.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ 1
Больной А., 67 лет. Госпитализирован в кардиологическое отделение Областной клинической больницы (Саратов) с жалобами на давящие боли за грудиной, отеки лица, живота, нижних конечностей, одышку при незначительной физической нагрузке, повышение артериального давления (АД) до 220 и 100 мм рт.ст., появившиеся за неделю до госпитализации после острой респираторной вирусной инфекции. Ранее заболеваний почек не было.
В ходе проведенного обследования данных в пользу острого коронарного синдрома получено не было. При эхокардиографии фракция изгнания сохранена. При обследовании впервые выявлен мочевой синдром (микрогематурия, протеинурия 3,0 г/л), повышение креатинина крови до 214 мкмоль/л. С учетом наличия клинико-лабораторных признаков острого нефритического синдрома (отеки, артериальная гипертензия, мочевой синдром) выставлен диагноз острого гломерулонефрита. Пациент переведен в нефрологическое отделение.
Состояние пациента тяжелое, обусловлено выраженным отечным синдром, тяжелой артериальной гипертензией. Температура тела нормальная. В легких жесткое дыхание, хрипов нет. Нижний край печени пальпируется на 2 см ниже реберной дуги. Селезенка не пальпируется. Выявлены анемия легкой степени (гемоглобин – 110 г/л), лейкоцитоз до 22,9 тыс./мкл с палочкоядерным сдвигом до 17%, тромбоцитоз до 645 тыс./мкл, единичные миелоциты, повышение уровня креатинина до 241 мкмоль/л, коагулограмма – гиперкоагуляция (АЧТВ – 35,4 с). В анализах мочи микрогематурия, протеинурия до 5,25 г/сут. При ультразвуковом исследовании выявлены гепатоспленомегалия, портальная гипертензия (v. lienalis – 9 мм, v. portae – 16 мм), при компьютерной томографии (КТ) – интерстициальные изменения легочной ткани с двух сторон, лимфаденопатия средостения.
С учетом клинической картины, гематологических изменений, гепатоспленомегалии высказано предположение о наличии онкогематологического заболевания. Гематологом установлен диагноз недифференцированного лимфопролиферативного заболевания. При иммунохимическом исследовании белков сыворотки крови и мочи моноклональной секреции не выявлено. В миелограмме (стернальная пункция) – нормоклеточный костный мозг. Рентгенографически костно-деструктивный изменений скелета не обнаружено.
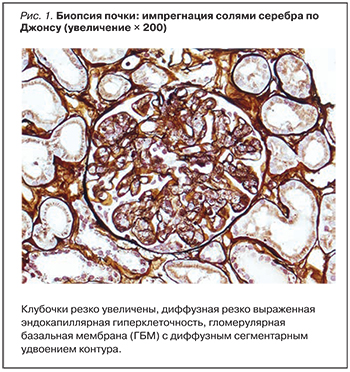 С целью морфологической верификации нефропатии выполнена диагностическая нефробиопсия, выявившая картину мембранопролиферативного гломерулонефрита (МПГН). Морфологическое исследование проводилось в ООО «Национальный центр клинической морфологической диагностики» Санкт-Петербурга.
С целью морфологической верификации нефропатии выполнена диагностическая нефробиопсия, выявившая картину мембранопролиферативного гломерулонефрита (МПГН). Морфологическое исследование проводилось в ООО «Национальный центр клинической морфологической диагностики» Санкт-Петербурга.
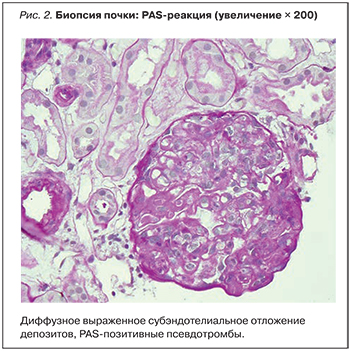
Световая микроскопия: 1 из 10 клубочков полностью склерозирован. Клубочки резко увеличены с диффузной, резко выраженной эндокапиллярной гиперклеточностью, диффузным, выраженным субэндотелиальным отложением депозитов с протрузией в просвет капилляров и формированием многочисленных PAS-позитивных псевдотромбов в просветах капилляров клубочков, диффузным сегментарным удвоением контура гломерулярной базальной мембраны (рис. 1, 2).
Заключение: гистологическая картина диффузной гломерулопатии с мембранопролиферативным поражением. Полный (1:10) и вторичный сегментарный (2:10) гломерулосклероз. Клинический диагноз: вторичный гломерулонефрит, мембранопролиферативный вариант в рамках недифференцированного лимфопролиферативного заболевания.
Через 2 нед отмечено ухудшение состояния пациента: усиление одышки, нарастание отечного синдрома, появление олигурии. Выявлено увеличение лейкоцитоза до 26 тыс./мкл, тромбоцитоза до 1088 тыс./мкл, ухудшение почечной функции (креатинин – 556 мкмоль/л), что потребовало проведения заместительной почечной терапии гемодиализом. Состояние пациента значительно улучшилось, одышка и отечный синдром купировались, диурез восстановился. Однако сохранялась гиперазотемия (креатинин – 629 мкмоль/л), лейкоцитоз до 43 тыс./мкл, тромбоцитоз до 1588 тыс./ мкл. К терапии добавлен преднизолон в дозе 15 мг/сут с последующим постепенным снижением дозы до полной отмены. В динамике отмечено разрешение острого почечного повреждения, снижение числа лейкоцитов до 12,2 тыс./мкл, тромбоцитов до 712 тыс./мкл.
 Повторное исследовании костного мозга (трепанобиопсия) выявило морфологическую картину ранней стадии ПМФ: гиперклеточность костного мозга с пролиферацией гранулоцитарного ростка, мегакариоцитарного ростка с атипией, скопление полиморфных мегакариоцитов. После консультации гематолога сформулирован диагноз «первичный хронический миелофиброз. Вторичный гломерулонефрит, мембранопролиферативный вариант».
Повторное исследовании костного мозга (трепанобиопсия) выявило морфологическую картину ранней стадии ПМФ: гиперклеточность костного мозга с пролиферацией гранулоцитарного ростка, мегакариоцитарного ростка с атипией, скопление полиморфных мегакариоцитов. После консультации гематолога сформулирован диагноз «первичный хронический миелофиброз. Вторичный гломерулонефрит, мембранопролиферативный вариант».
Начата терапия интерфероном альфа в дозе 9 млн ЕД./нед, антикоагулянтами продолжена 3-компонентная антигипертензивная терапия. На фоне 2-месячной интерферонотерапии отмечен удовлетворительный гематологический ответ – уменьшение тромбоцитоза и нормализация числа лейкоцитов. В конце июня 2016 г. появился выраженный отечный синдром, при стационарном обследовании выявлен нефротический синдром (НС): общий белок – 48 г/л, альбумины – 29 г/л, суточная протеинурия – 12,6 г. Начата терапия преднизолоном в дозе 30 мг/сут. В сентябре 2016 г. достигнута ремиссия НС, сохранялся стабильный гематологический ответ. В течение 2 лет состояние пациента остается удовлетворительным, продолжается терапия интерфероном альфа 9 млн ЕД./нед, антикоагулянтами, антигипертензивная терапия.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ 2
Больной Г., 60 лет. Госпитализирован в нефрологическое отделение в августе 2017 г. с жалобами на отеки лица, нижних конечностей, повышение АД до 150 и 100 мм рт.ст. В 2011 г. у него была диагностирована подагра, подагрический артрит, впервые отмечено повышение АД до 150 и 100 мм рт.ст. Уровень мочевой кислоты крови – до 550 мкмоль/л, анализы мочи – без патологии. Пациенту проводилось лечение аллопуринолом (200 мг/сут), антигипертензивная терапия. В мае 2017 г. появились отеки лица, нижних конечностей, выявлен мочевой синдром (гематурия, протеинурия до 1 г/л), расцененный как проявление подагрической нефропатии. При обследовании в нефрологическом отделении Областной клинической больницы (Саратов) выявлены клинико-лабораторные признаки НС: отеки, протеинурия 5,6 г/сут, гипо- и диспротеинемия (общий белок – 47 г/л, альбумин – 27 г/л); анемия (гемоглобин – 116 г/л), повышение уровня креатинина до 200 мкмоль/л.
В миелограмме плазматизации костного мозга не выявлено. Методом иммунотурбодиметрии выявлено повышенное содержание κ и λ свободных легких цепей (СЛЦ) иммуноглобулинов сыворотки крови без нарушения их соотношения (κ – 43,7 мг/л, λ – 46,7 мг/л, отношение κ/λ-СЛЦ – 0,94). При рентгенографии плоских костей скелета костно-деструктивных изменений не обнаружено. Диагностическая нефробиопсия (морфологическое исследование проводилось на кафедре патологической анатомии Первого МГМУ им. И.М. Сеченова) выявила картину МПГН («лапчатая» структура клубочков, умеренная мезангиальная пролиферация, утолщение базальной мембраны). Заключение: наиболее вероятной причиной МПГН является БОЛЦ. Таким образом, установлен диагноз «моноклональная гаммапатия. Хронический гломерулонефрит, мембранопролиферативный вариант (болезнь отложения легких цепей)».
С учетом высокой активности нефропатии начата программная терапия: метилпреднизолон по 1000 мг внутривенно в течение 3 дней, в дальнейшем преднизолон по 40 мг/сут. С ноября 2017 г. к лечению добавлен циклофосфамид в виде внутривенных инфузий по 1000 мг каждые 4 нед. В течение года сохранялся НС (протеинурия – 4,0 г/ сут, общий белок – 45 г/л, альбумины – 26 г/л). В ноябре 2018 г. отмечена незначительная положительная динамика в виде уменьшения выраженности отечного синдрома, снижения уровня протеинурии, повышения уровня протеинов сыворотки крови. Учитывая отсутствие разработанных рекомендаций по лечению БОЛЦ, в настоящий момент решается вопрос о ведении пациента в соответствии с принципами химиотерапии множественной миеломы.
ОБСУЖДЕНИЕ
ПМФ представляет собой клональный миелопролиферативный процесс, для которого характерно развитие фиброза костного мозга с появлением экстрамедуллярных очагов гемопоэза в результате злокачественной трансформации стволовой полипотентной клетки-предшественницы миелопоэза [1, 3, 8]. Наиболее часто заболевание дебютирует в возрасте 50–60 лет. Для течения ПМФ характерно наличие хронической фазы и фазы бластного криза, отражающей прогрессирование заболевания [3]. Наиболее характерными признаками ПМФ выступают спленомегалия и лейкоэритробластическая картина периферической крови [3, 8, 9].
Этиология заболевания неизвестна, в настоящий момент продолжается изучение молекулярно-генетических механизмов, лежащих в основе развития ПМФ [4, 10]. Открытие в последние годы новых генетических мутаций при ПМФ способствовало созданию новых таргетных препаратов – ингибиторов JAK2 (руксолитиниб) [8, 10, 11]. Тем не менее оптимальные схемы лечения больных не разработаны [8, 12]. Влияние препарата на общую выживаемость пациентов до конца не изучено [11, 12, 14]. Средняя продолжительность жизни при ПМФ составляет 6 лет. Единственным радикальным методом лечения остается аллогенная трансплантация костного мозга [3, 12]. Тактика лечения и выживаемость при ПМФ зависят от индивидуального прогноза пациента, рассчитанного по международной системе стратификации риска [3, 12–14].
В первом клиническом наблюдении продемонстрирован редкий случай развития нефропатии при ПМФ. У пациента в дебюте преобладали клинико-лабораторные симптомы гематологического заболевания (анемия, лейкоцитоз, тромбоцитоз, гепатоспленомегалия) в сочетании с острым повреждением почек. Вторичный генез нефропатии был заподозрен клинически. У пациента наблюдалось характерное для МПГН течение: острый нефритический синдром в дебюте заболевания, прогрессирующее снижение функции почек с последующим развитием нефротического синдрома. При этом, по данным литературы, поражение почек при ПМФ встречается крайне редко [5, 8, 9, 15]. Описано всего несколько случаев развития гломерулонефрита при ПМФ. W.Y. Au и соавт. (1999) и I. Kaygusuz и соавт. (2010) опубликовали 2 клинических наблюдения развития фокального сегментарного гломерулярного склероза у больных ПМФ [9, 15]. T.T. Liu и соавт. и M.A. Perazella и соавт. описали 2 пациентов с поражением почек в рамках ПМФ, у которых регистрировался нефротический синдром, при этом морфологически была выявлена картина мезангиопролиферативного гломерулонефрита [9, 15].
S.M. Said и соавт. (2011) продемонстрировали данные ретроспективного анализа историй болезни пациентов с поражением почек в рамках МПЗ [5]. Всего описано 11 случаев, в том числе 8 у больных ПМФ. У всех пациентов с ПМФ выявлена лейкоэритробластическая картина крови, анемия, протеинурия, повышение уровня креатинина, у двух больных регистрировался нефротический синдром. При морфологической верификации диагноза у всех пациентов выявлено удвоение контура гломерулярной базальной мембраны, признаки мезангиальной гиперклеточности, фиброза при световой микроскопии, что отмечалось и в приводимом наблюдении. У всех больных ПМФ диагностирована хроническая почечная недостаточность (ХПН), у 4 отмечалось развитие терминальной стадии ХПН.
Учитывая возраст и наличие симптомов опухолевой интоксикации, пациенту установлен промежуточный 1 риск по шкале IPSS [3, 14]. Для лечения таких пациентов препаратами первой линии симптоматической терапии являются циторедуктивные препараты (интерферон, гидроксимочевина) [3, 14]. У пациентов с более высоким риском для лечения используется таргетная терапия. Однако остановка начавшейся миелопролиферации и восстановление нормального кроветворения при ПМФ невозможны, что обусловливает отсутствие на фоне терапии гематологической ремиссии у пациентов.
Второе клиническое наблюдение демонстрирует развитие вторичного МПГН в рамках БОЛЦ. В клинической картине превалировало поражение почек с развитием тяжелого нефротического синдрома, почечной недостаточности, что характерно для БОЛЦ [7, 16]. В представленном наблюдении выявлена моноклональная секреция легких цепей. Отсутствие плазматизации костного мозга в миелограмме, морфологический профиль гломерулопатии позволили диагностировать БОЛЦ. Несмотря на проводимую иммуносупрессивную терапию, сохранялась высокая активность нефрита, обусловленная продолжающейся моноклональной секрецией.
БОЛЦ представляет собой системное заболевание; в основе его лежит пролиферация аномальных клонов плазматических клеток в костном мозге, продуцирующих избыточное количество моноклональных СЛЦ, которые откладываются в органах с последующим нарушением их функции [6, 16, 17]. Почки – ведущий орган-мишень при БОЛЦ [6, 16–18]. Это связано с тем, что циркулирующие СЛЦ подвергаются катаболизму путем клубочковой фильтрации, реабсорбции и эндоцитозу лизосомальными ферментами в клетках проксимальных канальцах [6]. Наиболее часто поражение почек проявляется в виде нефротического синдрома или изолированной протеинурии [7, 16, 17]. На момент постановки диагноза у 96% больных выявляют ХПН.
В настоящий момент рекомендации по терапии БОЛЦ отсутствуют [16]. Основу лечения составляет химиотерапия, направленная на снижение продукции парапротеинов. В большинстве случаев применяются программы химиотерапии множественной миеломы [16, 18, 19]. Продолжительность жизни больных зависит от степени нарушения вовлеченных в патологический процесс органов и варьируется от 1 до 10 лет [19].
ЗАКЛЮЧЕНИЕ
Особенностями данных клинических наблюдений является появление симптомов поражения почек в дебюте гематологического заболевания. В обоих случаях продемонстрированы трудности диагностики и лечения нефропатий в рамках гематологических заболеваний, обусловленные их редкой встречаемостью, недостаточной изученностью клинических проявлений, отсутствием разработанных и эффективных методов лечения. Представленные клинические наблюдения продемонстрировали необходимость исключения вторичного генеза нефропатии при морфологическом профиле МПГН при нефробиопсии.
