
ВВЕДЕНИЕ
Неалкогольная жировая болезнь печени (НАЖБП) – хроническое заболевание, характеризующееся избыточным накоплением нейтральных липидов в ≥5% гепатоцитов в отсутствие повреждающего действия алкоголя и других внешних причин [1]. По современным оценкам, различными формами НАЖБП страдают 25–30% мирового населения, что делает ее основной причиной хронической патологии печени [2]. Вследствие тесной связи метаболических изменений в организме с развитием НАЖБП в 2020 г. международным экспертным консенсусом был предложен новый термин, более точно учитывающий патогенез заболевания: метаболически ассоциированная жировая болезнь печени (МАЖБП) [3].
В последние годы, помимо широкого спектра метаболических и сердечно-сосудистых осложнений, все большее внимание специалистов привлекают неврологические нарушения, поведенческие, психические и когнитивные расстройства, ассоциированные с НАЖБП или неалкогольным стеатогепатитом (НАСГ). Особенный интерес ввиду сравнительно меньшей изученности представляют осложнения, сопровождающие прецирротические стадии заболевания, а также возможные поражения периферической нервной системы [4].
Хроническая болезнь печени и поражения центральной нервной системы (ЦНС) имеют широкий спектр общих метаболических факторов риска, включая малоподвижный образ жизни, ожирение, артериальную гипертензию, инсулинорезистентность, нарушения липидного обмена и альтерации состава кишечной микробиоты. Все перечисленные состояния не только ведут к возникновению и прогрессированию НАЖБП, но и способствуют развитию микро- и макроструктурных патологических изменений в нервной системе. Кроме этого, НАЖБП/НАСГ является самостоятельным фактором риска возникновения ряда цереброваскулярных, нейродегенеративных, когнитивных и психических расстройств [5, 6].
Согласно результатам систематического обзора данных доклинических исследований, все стадии НАЖБП/НАСГ ассоциированы с широким спектром органических и функциональных нарушений структур нервной системы. В их число входят нейровоспалительные реакции, нейродегенеративные процессы, эндотелиальная дисфункция, оксидативный стресс, микроангиопатия, ишемия и гипоксия ткани мозга, дисрегуляция основных систем нейротрансмиссии, геномные и рецепторные нарушения клеточного сигналинга, ослабление синаптической пластичности [4].
ПАТОГЕНЕЗ ПОРАЖЕНИЯ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ ПРИ НЕАЛКОГОЛЬНОЙ ЖИРОВОЙ БОЛЕЗНИ ПЕЧЕНИ
Нарушение обмена аммиака и гипераммониемия
Основным фактором развития центральных неврологических нарушений при НАЖБП принято считать токсическое воздействие эндогенного аммиака, обусловленное снижением эффективности его детоксификации в печени [7]. Основным путем нейтрализации аммиака у млекопитающих служит образование мочевины в орнитиновом цикле (цикле мочевины, ОЦ), протекающем в митохондриях и цитоплазме перипортальных гепатоцитов. Альтернативные механизмы детоксификации, имеющие значительно меньшее физиологическое значение, представлены амидированием глутамата и аспартата с последующим образованием глутамина и аспарагина соответственно, амидированием белков и восстановительным аминированием α-кетокислот с превращением пирувата в аланин, оксалоацетата – в аспарагин либо α-кетоглутарата (α‑КГ) – в глутамат [8].
При поражении печени вследствие репрессии ферментов ОЦ и угнетения синтеза орнитина de novo его внутренний пул уменьшается, что вызывает развитие гипераммониемии. Аммиак (в том числе в форме катиона аммония) поступает из системного кровотока в головной мозг путем пассивной диффузии через различные виды K+-каналов посредством Na+/K+-, Na+/H+-, Na+/K+/2Cl−- и K+/Cl−-котранспортеров, аквапоринов и Rh-гликопротеинов [9].
В поперечнополосатых миоцитах, перивенозных гепатоцитах и астроцитах происходит компенсаторная активация пути включения азота аммиака последовательно в глутамат и глутамин. ОЦ обеспечивает конверсию аммиака в мочевину в соотношении 2:1 с затратой 1,5 молекул аденозинтрифосфата (АТФ) с учетом дальнейшего включения фумарата в реакции окислительного фосфорилирования. В то же время синтез глутамина для утилизации одной молекулы аммиака требует гидролиза одной, а синтез аспарагина – двух макроэргических связей [8], что делает их существенно более энергозатратными. Чрезмерная активация этих путей сопряжена с развитием энергодефицита, приводящего к угнетению биосинтетических процессов в астроцитах, ослаблению антиоксидантной защиты и нарушению функциональной активности нейронов [10].
Нарушение энергетического обмена
Сдвиг метаболизма α‑КГ в сторону восстановительного аминирования избытком аммиака происходит в ущерб образованию интермедиатов цикла трикарбоновых кислот (ЦТК); следствием этого становится снижение аэробной энергопродукции и ингибирование многих синтетических процессов. Кроме этого, установлено, что α‑КГ принимает участие в активации сигналинга комплекса-1 механистической мишени рапамицина (mTORC1) – важнейшего медиатора инсулин-опосредуемых анаболических процессов, ингибитора аутофагии и положительного регулятора транскрипции, трансляции и клеточной пролиферации [11]. При ингибировании сигнальных путей mTORC1 нарушение протеостаза гепатоцитов влечет за собой истощение их белковых резервов и снижение синтетической функции печени, угнетение продукции фосфатидилхолина [12], активации кетогенеза [13], а также уменьшение клиренса липидов адипоцитами с развитием гипертриглицеридемии [14].
Дополнительным механизмом компенсации дефицита α‑КГ, а также гиперпродукции глутамина является трансаминирование последнего с пируватом под действием аланинаминотрансферазы (АЛТ). Несмотря на компенсаторную активацию гликолиза вследствие конверсии пирувата в лактат возможность его утилизации для аэробной энергопродукции значительно снижается. Часть образующегося при этом аланина в астроцитах снова превращается в лактат, используемый нейронами для генерации АТФ, а другая часть покидает ЦНС и выходит в системный кровоток [10, 15]. У пациентов с печеночной энцефалопатией (ПЭ) часто определяются повышенные концентрации лактата не только в сыворотке крови, но и в микродиализате, ликворе и головном мозге [10]. По данным доклинических исследований, возрастание уровня лактата способствует повышению внутричерепного давления вследствие отека мозга, а также нарушению энергетического баланса и гибели нейронов и глиальных клеток [16]. Тем не менее в связи с ценностью лактата как энергетического субстрата его дефицит может играть и негативную роль в течении ПЭ [17].
В соответствии с альтернативной точкой зрения ингибирование энергетического обмена в нейронах и глиальных клетках при ПЭ выступает следствием угнетения их функциональной активности, которая, в свою очередь, обусловлена повышением тонуса ГАМК-ергической системы на фоне гиперпродукции глутамина [18]. Накопление лактата в тканях ЦНС при этом объясняется не только активацией его продукции, но и снижением скорости расходования нейронами [19].
Нарушение обмена глутамина
Гиперпродукция глутамина и дисрегуляция цикла глутамата/глутамина/гамма-аминомасляной кислоты (ГАМК) – не менее важный фактор поражения ЦНС при различной патологии печени, чем собственно гипераммониемия [9, 20, 21]. Значительная часть глутамина может транспортироваться в митохондриальный матрикс и подвергаться глутаминолизу с обратным высвобождением аммиака. Увеличение pH среды при этом приводит к переходу проницаемости внутренней митохондриальной мембраны, исчезновению мембранного электрического градиента, осмотическому набуханию митохондрий, развитию митохондриальной дисфункции и генерации активных форм кислорода (АФК). Глутамин обладает собственной осмотической активностью и дополнительно усугубляет набухание и дистрофическую трансформацию астроцитов, тем самым способствуя развитию цитотоксического отека головного мозга и повышению внутричерепного давления [20, 21].
Избыток глутамина также подвергается метаболизму почечной и кишечной глутаминазами с высвобождением уже двух молекул аммиака, что формирует порочный круг [22]. Часть образуемого при этом глутамата может дезаминироваться под действием глутаматдегидрогеназы (ГДГ) с образованием α‑КГ и еще одной молекулы аммиака, причем из-за низкой аффинности ГДГ к последнему протекание обратной реакции затруднено [23].
Токсические концентрации аммиака нарушают эффекторные пути различных подтипов рецепторов глутамата (NMDAR-I, AMPAR, mGluR) и опосредуемые глутаматом нейрон-астроцитарные взаимодействия. Гипераммониемия вызывает избыточную активацию NMDAR, кальциневрина и Na+/H+-АТФазы, а также угнетение продукции кинуреновой кислоты, что сопровождается нарушением цитоплазматического и митохондриального гомеостаза кальция, развитием энергетического дефицита, митохондриальной дисфункции и оксидативного стресса [24, 25].
Нейростероидогенез и дисрегуляция цикла глутамин/глутамат/ГАМК
Помимо аммиака, центральной нейротоксичностью обладают меркаптаны, фенолы, короткоцепочечные жирные кислоты, марганец, ложные нейротрансмиттеры (тирамин, октопамин, β-фенилэтаноламин) и эндогенные модуляторы активности рецепторов ГАМК (нейростероиды, диазепам, нордиазепам, эндозепины). Интенсивная конверсия глутамата в глутамин существенно ограничивает скорость продукции ГАМК, что наряду с блокадой астроцитарных транспортеров этого соединения приводит к сенситизации ГАМКА-рецепторов и ассоциированных сайтов связывания к воздействию всех эндогенных агонистов и положительных аллостерических модуляторов [24, 26, 27].
Установлено, что гипераммониемия способствует увеличению числа глиальных белков-транслокаторов (18 kDa) (TSPO), ранее известных как периферические бензодиазепиновые рецепторы, а также повышает аффинность некоторых лигандов к сайтам связывания, сопряженным с TSPO. По данным исследований in vitro и in vivo, как гипераммониемии, так и введению экзогенных агонистов TSPO сопутствует развитие оксидативного и нитрозативного стресса, что, в свою очередь, нарушает функциональную активность глиальных клеток и вызывает гиперэкспрессию NMDAR [28]. В сочетании с прямым увеличением мембранного потенциала, сенситизацией NMDAR и нарушением сигналинга циклического гуанозинмонофосфата (цГМФ) под влиянием аммиака активация TSPO способствует эксайтотоксическому поражению нейронов и утрате нейрональной пластичности [25].
Поскольку TSPO опосредует захват холестерина, используемого для биосинтеза нейростероидов, аммиак, наряду с ионами марганца и провоспалительными цитокинами, косвенно индуцирует продукцию основных представителей этой группы соединений – прегненолона, аллопрегнанолона (ALLO) и тетрагидродезоксикортикостерона (THDOC). ALLO и другие нейростероиды, повышающие тонус ГАМК-ергической нейротрансмиссии, угнетают процессы синаптической передачи и долговременной потенциации, что вносит значительный вклад в развитие когнитивного и мнестического дефицита, а также нарушения цикла сна/бодрствования [29].
Кроме этого, при хронической ПЭ наблюдается угнетение биосинтеза еще одного нейростероида – дегидроэпиандростерона сульфата (DHEAS), обладающего отрицательным модулирующим эффектом в отношении ГАМКА-рецепторов [29]. Предположительно, дисрегуляция нейростероидогенеза на фоне гипераммониемии может также оказывать отрицательное воздействие на серотонин-, холин-, глицин- и опиоидергическую системы на геномном и рецепторном уровнях [29, 30].
Центральная инсулинорезистентность
Снижение чувствительности паренхимы головного мозга к инсулину, тесно взаимосвязанное с НАЖБП, в наибольшей степени обусловлено накоплением липидов в клетках печени, развитием хронического воспалительного ответа и стресса эндоплазматического ретикулума (ЭПР). Избыточные количества диацилглицеридов и церамидов в гепатоцитах блокируют сигнальные пути инсулина на уровне его рецептора, в то время как провоспалительные цитокины и медиаторы стресса ЭПР активируют c-Jun N-терминальные киназы (JNK), опосредующие отрицательную обратную связь с инсулиновым рецептором [31]. Наибольшая чувствительность к действию инсулина свойственна нейронам и глиальным клеткам коры, гиппокампа, гипоталамуса и мозжечка [32].
Хроническое повышение уровней глюкозы и свободных жирных кислот (СЖК) в крови приводит к дисфункции эндотелиальных клеток гематоэнцефалического барьера (ГЭБ), нарушению его целостности и проницаемости для различных веществ, включая эндогенные токсические соединения, инсулин, провоспалительные цитокины и сами СЖК. Возрастание концентраций СЖК в ЦНС, в том числе вследствие высвобождения их астроцитами, способствует активации микроглии, развитию инсулинорезистентности, местной воспалительной реакции и оксидативного стресса, формируя тем самым порочный круг [32].
Центральная инсулинорезистентность, наблюдаемая у животных с экспериментальной минимальной печеночной энцефалопатией (МПЭ), вызывает снижение продукции нейротрофических факторов, дисрегуляцию сигнальных путей рецепторов N-метил-D-аспартата (NMDAR) [33], mTORC1, киназы гликогенсинтазы-3β и митоген-активируемых протеинкиназ (MAPK). Перечисленные изменения приводят к нарушению аутофагии и нормальной деградации белков с аномальной структурой, пролиферации нейрональных клеток-предшественников, нейрональной поляризации и пластичности, организации цитоскелета и энергетического метаболизма нейронов [32]. Наряду с этим развитие инсулинорезистентности паренхимы головного мозга сопровождается нарушением функции митохондрий, интенсификацией перекисного окисления липидов и белков в областях стриатума и n. accumbens, а также экспрессией аберрантных форм моноаминооксидазы. Резкое угнетение обмена дофамина в мезолимбическом и нигростриарном трактах становится причиной развития у грызунов поведенческих нарушений, которые коррелируют с тревожными и депрессивными расстройствами у пациентов с ПЭ [34].
Нарушение кишечного микробиома
Нарушение кишечного микробиома при НАЖБП главным образом характеризуется изменением количественного соотношения между филами Firmicutes (в частности, семейства Lactobacillaceae), Bacteroidetes и Proteobacteria. Увеличение численности грамотрицательных бактерий в сочетании с патологическим повышением проницаемости кишечного барьера сопровождается избыточной продукцией нейротоксичных соединений (аммиака, этанола, фенола), липополисахаридов и других эндотоксинов. Перечисленные продукты микробного синтеза стимулируют развитие системного и локального воспалительного ответа, оксидативного стресса, эндотелиальной дисфункции и нейродегенеративного поражения ЦНС [35, 36].
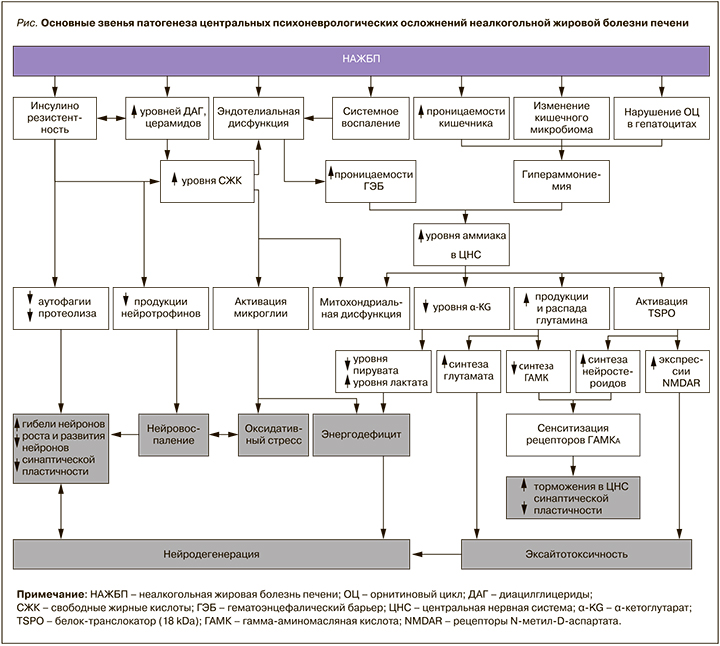
Основные звенья патогенеза центральных психоневрологических осложнений НАЖБП представлены на рисунке.
ПЕЧЕНОЧНАЯ ЭНЦЕФАЛОПАТИЯ
Нейропсихиатрические нарушения, сопутствующие острым и хроническим поражениям печени различной этиологии, принято объединять в синдром печеночной (портосистемной) энцефалопатии (ПЭ) – потенциально обратимое нарушение психомоторной, интеллектуальной, эмоциональной и поведенческой функций ЦНС [7, 37].
Согласно критериям West–Haven (WHC), в течении ПЭ выделяют минимальную стадию (МПЭ) и 4 последовательные стадии явной ПЭ (ЯПЭ) [37]. Классификация Международного общества по изучению ПЭ и метаболизма азота объединяет МПЭ и I стадию в латентную ПЭ (ЛПЭ), а II–IV стадии – в ЯПЭ. Тип А соответствует МПЭ или ПЭ I стадии (ЛПЭ), вызванной острой печеночной недостаточностью. Тип В обусловлен главным образом портосистемным шунтированием и включает II и III стадии ЯПЭ; тип C соответствует ЯПЭ IV стадии на фоне цирроза печени любой этиологии [37].
Психометрическое тестирование больных с МПЭ позволяет выявить ранние, субклинические нарушения зрительно-пространственных функций и абстрактного мышления, замедление психомоторных реакций, ухудшение мелкой моторики и конструктивную апраксию. По мере прогрессирования ПЭ возникают апатия, депрессия, расстройства личности и поведения, дезориентация во времени и пространстве, нарушения цикла сон/бодрствование [38].
Риск перехода ПЭ в манифестную форму значительно увеличивается из-за дальнейшего снижения эффективности детоксикации венозной крови как при спонтанной коллатерализации кровотока на фоне портальной гипертензии, так и после формирования искусственных портосистемных анастомозов хирургическим путем. После проведения трансъюгулярного внутрипеченочного портосистемного шунтирования (TIPS) ЯПЭ впервые развивается у 20–70% больных, при этом у 8% из них она является рефрактерной к лечению [39, 40]. При выполнении дистального спленоренального шунтирования частота манифестации ПЭ составляет 55–67% [41].
Для поздних стадий ЯПЭ характерно развитие психотических состояний, делирия, прогрессирующей дезориентации во времени и пространстве, острого угнетения сознания от сонливости до сопора и комы. Двигательные нарушения включают расстройства речи и мимики, атаксию, диспраксию, астериксис («порхающий тремор»), пирамидные (гиперрефлексия, положительный рефлекс Бабинского) и экстрапирамидные симптомы (мышечная ригидность, брадикинезия, постуральная недостаточность) [37]. Помимо вышеперечисленных симптомов, для ПЭ характерны расстройства сна, пищевого поведения и сексуальной активности [7, 42].
ПРИОБРЕТЕННАЯ (НЕВИЛЬСОНОВСКАЯ) ГЕПАТОЦЕРЕБРАЛЬНАЯ ДЕГЕНЕРАЦИЯ
У небольшой части больных неалкогольным циррозом печени развивается приобретенная (невильсоновская) гепатоцеребральная дегенерация (ПНГД), патогенетически связанная с наличием портальной гипертензии и характеризующаяся необратимым поражением головного мозга вследствие нарушений метаболизма аммиака, ароматических аминокислот и тяжелых металлов, главным образом марганца. Накопление марганца вследствие недостаточной элиминации с желчью приводит к нарушению энергетического метаболизма глиальных клеток и их апоптозу, что обусловливает развитие токсической нейродегенерации преимущественно в областях бледного шара, стриатума и ретикулярной части черной субстанции [43, 44].
Симптоматика ПНГД представлена сочетанием когнитивных, эмоциональных и экстрапирамидных расстройств. Чаще всего отмечаются акинезия, мышечная ригидность, тремор, миоклонус, апатия, летаргия, депрессивные состояния, снижение внимания, памяти и интеллекта, иногда с развитием вторичной деменции. К поздним осложнениям ПНГД относятся миелопатия с симптомами медленно прогрессирующего нижнего спастического парапареза, нарушениями чувствительности и расстройствами функции тазовых органов, а также полинейропатия по демиелинизирующему типу [43, 44].
НЕЙРОДЕГЕНЕРАТИВНЫЕ ПРОЦЕССЫ
Системное воспаление и активация CD4+-Т-лимфоцитов при НАСГ сопровождаются инфильтрацией ими мозговых оболочек и межоболочечных пространств, что вызывает рекрутирование, миграцию и поляризацию по М1-фенотипу микроглиальных клеток, продуцирующих провоспалительные цитокины и хемокины [45, 46]. Нарушения липидного обмена, дисфункция антиоксидантных систем и стресс ЭПР сопровождаются накоплением в паренхиме головного мозга липидных капель, а это дополнительно стимулирует активацию микроглии и развитие нейровоспалительных реакций. Избыточное накопление липидных капель ассоциировано с рядом нейродегенеративных заболеваний, таких как боковой амиотрофический склероз, болезнь Альцгеймера, болезнь Паркинсона, хорея Гентингтона и наследственная спастическая параплегия [47].
У пациентов с ранними стадиями НАСГ при исследовании post mortem обнаруживались множественные признаки дегенерации мозжечка, включая гиперактивацию микроглии и астроцитов молекулярного слоя коры, лимфоцитарную инфильтрацию, гибель клеток Пуркинье и гранулярных клеток, нарушение микроциркуляции и образование микротромбов [45, 46]. По мере прогрессирования основного заболевания нейровоспалительные и нейродегенеративные процессы затрагивают подлежащие слои коры и белое вещество мозжечка как одно из наиболее уязвимых структур [48] и впоследствии распространяются на другие регионы головного мозга (45).
Гипераммониемия и повышение проницаемости ГЭБ для аммиака на фоне НАЖБП связаны со многими факторами патогенеза болезни Альцгеймера, в том числе с накоплением β-амилоида, дистрофией и гибелью глиальных клеток и нейронов, ишемией головного мозга, митохондриальной дисфункцией, оксидативным стрессом, хроническим нейровоспалением, инсулинорезистентностью, утратой синаптической пластичности и гиперактивацией ГАМК-опосредуемых процессов торможения [49]. Отмечается, что характер астроцитоза при ПЭ схож с таковым при болезни Альцгеймера и других нейродегенеративных процессах. Так называемые астроциты Альцгеймера II типа выявляются у многих или даже у всех пациентов с ПЭ и характеризуются резким увеличением ядра со смещением к периферии хроматина, накоплением включений гликогена, увеличением эндоплазматического ретикулума и митохондрий [50].
Прогрессирование НАЖБП ассоциировано с усугублением течения такого наследственного заболевания, как атаксия-телеангиэктазия, и его неврологических проявлений. Это связывают с ускорением процесса нейродегенерации вследствие хронического системного воспаления, оксидативного стресса и атеросклеротического поражения сосудов [51]. В качестве важнейшего общего звена патогенеза как НАЖБП, так и атаксии-телеангиэктазии и болезни Альцгеймера рассматривается нарушение активности киназы АТМ (ataxia-telangiectasia mutated) и ассоциированных процессов повреждения ДНК и апоптоза нейронов [52, 53], однако характер взаимосвязи патологий печени и дисрегуляции АТМ остается малоизученным [54].
Установлено, что наличие НАЖБП и/или НАСГ способствует накоплению и отложению β-амилоида в тканях головного мозга, что обусловлено ухудшением связывания этого вещества с альбуминами крови и его печеночного клиренса, а также повышением проницаемости ГЭБ [55]. В исследовании на мышах было показано, что алиментарная НАЖБП способствует развитию не только различных форм болезни Альцгеймера, но и церебральной амилоидной микроангиопатии, характеризующейся отложением β-амилоида в стенке (медии и адвентиции) артерий среднего и малого диаметра [55].
С использованием нейросетевых методов кластеризации и анализа белковых взаимодействий было выделено 189 генов, предположительно общих для НАЖБП и нейродегенерации, регулирующих сигналинг интерлейкина 17 (ИЛ-17), а также обмен углеводов и длинноцепочечных жирных кислот [57]. Обнаружено, что тяжесть неалкогольного фиброза печени прямо коррелирует с наличием и распространенностью поражения белого вещества головного мозга [58], расцениваемого как одно из морфологических проявлений болезни Альцгеймера [59].
Тем не менее данные клинических исследований о взаимосвязи НАЖБП/НАСГ и собственно болезни Альцгеймера несколько противоречивы. В когортном исследовании с участием 656 пациентов наличие НАЖБП само по себе не было значительно ассоциировано с заболеваемостью болезнью Альцгеймера (3,3 против 4,9% в контрольной группе), однако тяжесть гистологического повреждения, в частности наличие фиброза печени, имела существенную предиктивную ценность для оценки риска развития болезни Альцгеймера в дальнейшем [60]. По данным ретроспективного анализа данных Биобанка Великобритании (n≈455 226), НАЖБП на стадии фиброза печени выступает независимым фактором риска развития сосудистой деменции и деменции в целом (отношение рисков (ОР) 3,45), однако корреляция между заболеваемостью болезнью Альцгеймера и наличием фиброза печени (ОР 1,24) имела пограничную статистическую значимость [61].
Взаимосвязь между НАЖБП и болезнью Паркинсона имеет гендерные различия: риск этого тяжелого неврологического заболевания снижен у мужчин (ОР 0,86), но повышен у женщин с патологией печени (ОР 1,09). Кроме этого, статистически значимая корреляция прослеживается у пациентов 40–65 лет, но практически отсутствует в более пожилом (≥65 лет) возрасте [62].
Распространенность стеатоза печени среди больных боковым амиотрофическим склерозом значительно превышает таковую в общей популяции (76 против ≈25%), что может указывать на наличие общих факторов нарушения липидного обмена при обоих заболеваниях [63]. По некоторым предположениям, гипераммониемия при НАЖБП в сочетании с наблюдаемым при боковом амиотрофическом склерозе дефицитом защитных кальций-связывающих белков (кальбиндина, кальретикулина, парвальбумина) может опосредовать повреждение и гибель двигательных нейронов [64].
ЦЕРЕБРОВАСКУЛЯРНЫЕ НАРУШЕНИЯ
Гиперпродукция провоспалительных цитокинов и ростовых транскрипционных факторов, а также дисрегуляция секреции адипокинов способствуют развитию эндотелиальной дисфункции и дополнительно усугубляют атеросклеротическое поражение сосудов при дислипидемии [6, 65]. Хроническое воспаление и нарушение синтетической функции печени также влекут за собой повышение активности тромбоцитов и нарушение реологических параметров крови с формированием гиперкоагуляционного состояния. Установлено, что у пациентов с различными стадиями НАСГ наблюдается индукция ингибитора активации плазминогена-1, С-реактивного белка, фибриногена, факторов свертывания крови III, VII, VIII, IX, XII, уменьшение клиренса и накопление фактора фон Виллебранда, а также снижение продукции антитромбина [66, 67]. ИЛ-1, ИЛ-6, фактор некроза опухоли-альфа (ФНО-α) и другие провоспалительные цитокины стимулируют синтез тромбопоэтина гепатоцитами, дифференцировку тромбоцитов и увеличение их в размерах с соответственным повышением продукции прокоагуляционных факторов [67].
С прогрессированием НАЖБП до стадии цирроза, ухудшением протеинсинтетической функции печени и тромбоцитопении вследствие вторичной спленомегалии происходит обратный сдвиг равновесия систем свертывания крови в сторону гипокоагуляции с развитием хронического геморрагического синдрома. Тем не менее ввиду истощения резерва как про-, так и антикоагуляционных факторов баланс системы гемостаза становится крайне неустойчив, при тяжелых поражениях печени вне зависимости от этиологии могут возникать не только кровотечения, но и тромбозы [68].
Церебральная микроангиопатия на фоне НАСГ сопряжена с повышенной частотой возникновения как микротромбов, так и бессимптомных церебральных микрогеморрагий, что приводит к образованию очагов глиоза в белом веществе [46, 69]. Наличие НАЖБП служит независимым фактором риска возникновения геморрагического [70], а также лакунарного [71] и иных видов ишемического инсульта, предиктором его тяжести, степени инвалидизации, краткосрочной и отсроченной смертности [72, 73]. Кроме этого, тяжесть неалкогольного фиброза печени положительно коррелирует с вероятностью геморрагической трансформации инфаркта головного мозга [74].
ПСИХИЧЕСКИЕ РАССТРОЙСТВА
В Диагностическом и статистическом руководстве по психическим расстройствам 5-го издания (Diagnostic and statistical manual of mental disorders; DSM-5) отмечаются психотические расстройства, делирий, кататонический синдром, большие и малые нейрокогнитивные расстройства как возможные варианты вторичных нарушений, вызванных соматической патологией, в том числе наблюдаемых на фоне патологии печени [75].
Согласно имеющимся данным, наиболее распространенным видом нарушений психики у больных НАЖБП на прецирротических стадиях являются аффективные расстройства. Так, у 53% пациентов обнаруживаются признаки субклинической депрессии, а у 14% – клинической [76]. Заболеваемость депрессией находится в прямой корреляции с выраженностью стеатоза печени и значением индекса FLI (Fatty Liver Index) [77, 78]. Женщины, имеющие стеатоз печени, более подвержены депрессии в сравнении с мужчинами [78]; кроме пола, достоверно известными факторами риска депрессивных расстройств при НАЖБП служат ожирение, сахарный диабет 2-го типа, курение и/или наличие заболеваний легких [78].
Пациенты с НАСГ характеризуются значительно более высоким риском развития хотя бы одного эпизода большого депрессивного расстройства (ОР 3,8) и/или генерализованного тревожного расстройства (ОР 5,0) в течение жизни [80] по сравнению как со здоровыми людьми, так и имеющими стеатоз печени без воспалительной реакции [79]. Предположительно взаимосвязь между НАЖБП/НАСГ и депрессивными расстройствами носит двусторонний характер: наличие депрессии может усугублять течение патологии печени, в том числе из-за необходимости проведения длительной терапии гепатотоксичными психотропными лекарственными средствами, а также возможного изменения образа жизни и снижения приверженности к терапии. Вероятность адекватного ответа на проводимое лечение среди коморбидных пациентов оценивается также ниже, чем у лиц, не страдающих аффективными расстройствами [81, 82].
Тревожное расстройство в субклинической и клинической формах выявляется у 45 и 25% больных НАЖБП/НАСГ соответственно [76]. При этом вероятность формирования как реактивной, так и личностной тревожности достоверно повышена у женщин (ОР 1,84 и 2,45 соответственно), тогда как среди мужчин эта зависимость остается на уровне тенденции [78].
НАЖБП достоверно ассоциирована с большей частотой выявления расстройств личности в сравнении как с общей популяцией, так и с больными другими хроническими заболеваниями печени. Кроме этого, некоторые расстройства пищевого поведения (компульсивное переедание) значительно более тесно связаны с НАЖБП, нежели с другими поражениями органа [83]. Согласно предположениям, в число психических осложнений НАЖБП может также входить хроническое стрессовое расстройство, обусловленное психосоциальным стрессом [84].
КОГНИТИВНЫЕ И МНЕСТИЧЕСКИЕ НАРУШЕНИЯ
Есть основания полагать, что те или иные когнитивные нарушения осложняют течение НАЖБП практически у всех больных, половина из которых испытывает их в легкой форме, а другая половина – в среднетяжелой или тяжелой [85]. К ранним признакам когнитивного дефицита, чаще всего выявляемым при НАЖБП/НАСГ, относятся ослабление гиппокамп-зависимых видов памяти, нарушение визуального восприятия, исполнительных функций и способности к абстрактному мышлению [86, 87]. По данным доклинических исследований, ослабление краткосрочной и долгосрочной зрительно-пространственной памяти, ее консолидации и ретенции может быть обусловлено снижением метаболической активности нейронов гиппокампа, таламуса, энторинальной коры, миндалевидного и маммиллярных тел, а также снижением уровней норадреналина в стриатуме и дофамина в префронтальной коре и мозжечке [35].
В двух кросс-секционных популяционных исследованиях (44 и 213 человек) были получены данные о развитии у пациентов с НАЖБП дефицита зрительно-пространственной памяти согласно шкалам MoCA (Montreal cognitive assessment – Монреальская когнитивная шкала) и RBANS (repeatable battery for the assessment of neuropsychological status – повторяемая батарея оценки нейропсихологического статуса) [88, 89]. В исследовании типа «случай–контроль» (n=208) результаты теста повторения линий (line tracing test, LTT), используемого для оценки зрительно-пространственных функций при МПЭ, были нормальными у пациентов со стеатозом печени, но значительно сниженными в подгруппах, имевших НАЖБП на стадиях стеатогепатита, фиброза или цирроза печени [90]. Отмечается, что нарушения данного вида памяти могут выявляться при неалкогольном циррозе печени и в отсутствие симптомов ЯПЭ [91].
АСТЕНИЯ И РАССТРОЙСТВА СНА
До 70% больных различными стадиями НАЖБП страдают от повышенной утомляемости и хронической усталости, что отрицательно сказывается на уровне физической активности, снижает качество жизни, а также способствует формированию аффективных и когнитивных расстройств [92–94]. Когнитивно-мнестические нарушения, включая снижение внимания и усвоения новой информации, в сочетании с вегетативными симптомами могут рассматриваться как признаки неспецифического астенического синдрома. При этом отсутствие корреляций между когнитивной дисфункцией, биохимическими и гистологическими маркерами повреждения печени подтверждает, что проявления астении на ранних стадиях НАЖБП не связаны с наличием ПЭ [95, 96].
Астения, развивающаяся при НАЖБП, ухудшает качество жизни, профессиональную и повседневную деятельность пациентов [97]. Нередко она сочетается с когнитивно-мнестическими нарушениями, аффективными расстройствами (чаще всего тревожно-депрессивного спектра), вегетативными нарушениями и дневной сонливостью [96, 98].
Аномальная дневная сонливость может возникать как вследствие хронической усталости [96], так и являться самостоятельным осложнением прецирротической НАЖБП с распространенностью около 25% [99]. В число нарушений сна, ассоциированных с этим заболеванием, входят также бессонница (ОР 2,17), обструктивное апноэ во сне (ОР 1,15–1,39) и синдром беспокойных ног (ОР 1,16) [100, 101]. Согласно результатам прохождения Питтсбургского опросника на определение индекса качества сна (PSQI) и оценкам по Шкале сонливости Эпворта (ESS), доля пациентов с НАЖБП, страдающих существенными нарушениями сна, составляет 30,2% при отсутствии МПЭ и 74,2% при ее наличии. МПЭ достоверно ассоциирована с увеличением латентного времени сна, времени пробуждения и числа ночных пробуждений, а также ухудшением общего качества сна. В свою очередь, определение качества сна по опросникам PSQI и ESS, оценка времени пробуждения, относительной эффективности сна, латентного времени сна и частоты ночных пробуждений позволяют с высокой вероятностью (86,8%) предсказывать наличие МПЭ [102].
ПЕРИФЕРИЧЕСКАЯ И АВТОНОМНАЯ НЕЙРОПАТИЯ
Периферическим неврологическим осложнениям НАЖБП на сегодняшний день уделяется сравнительно меньшее внимание, нежели центральным. Считается, что поражение периферических нервов при НАСГ может быть связано с наличием инсулинорезистентности, дислипидемии и явлений липотоксичности, хронического системного воспаления и эндотелиальной дисфункции [103, 104].
Оксидативный стресс приводит к разобщению эндотелиальной NO-синтазы, развитию эндотелиальной дисфункции и расстройству эндоневрального кровотока, нарушению проводимости нервного волокна, аксональной атрофии и поражению волокон наименьшего диаметра типа A-δ и C (нейропатии малых волокон) [105]. ИЛ-1β, ИЛ-6, ИЛ-8, ФНО-α, интерферон-гамма, простагландин E2, CC- и CXC-хемокины опосредуют воспалительную нейродегенерацию и уменьшение скорости проведения электрического импульса, а также играют центральную роль в переходе безболевой нейропатии в болевую форму. Нарушение толерантности к глюкозе, гиперинсулинемия и инсулинорезистентность связаны с гиперпролиферацией клеток эндотелия, утолщением базальной мембраны и сужением просвета эндоневральных сосудов, что в сочетании с вазоконстрикцией и нарушением реологических параметров крови вызывает хроническую ишемию нервного волокна [105, 106].
Матриксные металлопротеиназы-2 и -9, экспрессия которых положительно коррелирует с активностью НАСГ и тяжестью фиброза [107, 108], могут нарушать баланс антероградной (Валлеровой) дегенерации аксонов и миелиновых оболочек дистальнее места повреждения нерва и их последующей регенерации. Предположительно ингибирование роста и регенерации аксонов может также быть ассоциировано с индукцией сигналинга mTOR и патологическим увеличением активности белков семейства семафоринов [105].
SARM1 (стерильный белок-1, содержащий мотивы α- и Toll/интерлейкинового рецептора), один из эффекторов ФНО-α, индуцирует гидролиз NAD+ и антероградную кальпаин-опосредованную дегенерацию аксонов центральных и периферических нейронов. Установлено, что активация сигнальных путей SARM1 лежит в основе печеночной симпатической нейропатии [109], а также поражения нервных волокон при травматических поражениях головного мозга, токсическом действии химиотерапевтических средств и нейродегенеративных заболеваниях, в том числе при болезни Альцгеймера, болезни Паркинсона и хорее Гентингтона [110].
В соответствии с данными клинических исследований минимальные признаки поражения периферических нервов выявляются электрофизиологическими методами у 53–73% пациентов с циррозом печени различной этиологии; у 29–52% отмечаются симптомы нейропатии [111–113]. Наиболее частым вариантом при циррозе печени является симметричная диффузная сенсомоторная полинейропатия с преобладанием поражения по аксональному типу, приблизительно в 2/3 случаев сочетающаяся с автономной нейропатией [111, 112].
В исследовании, включившем 4974 пациента с НАЖБП, часть из которых имела также сахарный диабет 2-го типа, признаки дисфункции автономной нервной системы, косвенно указывающие на наличие автономной нейропатии, были обнаружены в 40,8% случаях (ОР 1,38 по сравнению с контролем) [114]. Точная распространенность периферической и автономной нейропатии среди больных изолированной НАЖБП на прецирротических стадиях неизвестна.
ЗАКЛЮЧЕНИЕ
Результаты последних клинических исследований свидетельствуют о чрезвычайно высокой распространенности среди больных различными стадиями НАЖБП органических и функциональных расстройств ЦНС, которые негативно влияют на прогрессирование и прогноз в отношении основного заболевания, риск возникновения иных осложнений, качество жизни пациентов и эффективность проводимой терапии. Патогенез указанных расстройств достаточно сложен и включает нарушения детоксификации эндогенного аммиака, энергетического обмена, обмена глутамина, глутамата и ГАМК, баланса возбуждения и торможения в ЦНС, оксидативный и нитрозативный стресс, центральную инсулинорезистентность и множество других факторов.
Наиболее часто в клинической практике у больных прецирротической НАЖБП выявляются тревожно-депрессивные расстройства, признаки когнитивной и мнестической дисфункции, хроническая усталость, астения и расстройства сна. Неалкогольный цирроз печени в большей степени ассоциирован с комплексными токсическими и дегенеративными поражениями ЦНС, объединяемыми в синдром ПЭ. Несмотря на существенно возросшее в последние годы внимание специалистов к проблеме, распространенность, патогенетические и клинические аспекты, а также потенциальные подходы к фармакотерапии перечисленных состояний требуют дальнейшего детального изучения.


