Синдром Элерса–Данло (синонимы: синдром Черногубова, синдром Meekeren–Ehlers–Danlos, эластическая фибродисплазия, генерализованная фиброэластическая дисплазия, врожденная мезодермальная дистрофия, мезенхиматоз, гиперэластичность кожи) представляет собой гетерогенную группу системных наследственных заболеваний, обусловленных мутациями в генах коллагена, с клиническими проявлениями в виде гиперрастяжимости кожи, гипермобильности суставов, хрупкости тканей, изменениями сердечно-сосудистой и других систем организма [2, 3]. Синдром Элерса–Данло (СЭД) относится к моногенным заболеваниям с разными типами наследования, включающими аутосомно-доминантный, аутосомно-рецессивный и Х-сцепленный.
КЛАССИФИКАЦИЯ
В 1988 г. были впервые систематизированы знания и разработаны диагностические критерии СЭД (Берлинские критерии), включавшие 11 подтипов заболевания с большими и малыми критериями [4]. В 1998 г. новые результаты биохимических и молекулярных исследований, клинических симптомов позволили пересмотреть старую классификацию, описав 6 подтипов СЭД (Вильфраншские критерии) [5].
Наконец, за истекшие 2 десятилетия после опубликования Вильфраншских критериев появились новейшие наблюдения, обосновывающие клинические и генетические признаки 13 подтипов СЭД (табл. 1) [6].

РАСПРОСТРАНЕННОСТЬ
Распространенность СЭД составляет около 1:5000 популяции для всех типов, при этом гипермобильный тип встречается в половине всех зарегистрированных случаев. У лиц различных национальностей отмечают вариативность показателя распространенности гипермобильного типа: в кавказской популяции частота составляет 0,2–0,6%, у африканцев еще выше. Соотношение мужчин и женщин с СЭД составляет 1:8. Классический тип СЭД встречается 1 на 20 000–40 000 населения. Сосудистый тип встречается редко, данные о распространенности меняются, считают, что этот тип характерен для 1 из 250 тыс. человек [4, 5].
Артрохалазия и кифосколитический тип описаны менее чем у 100 человек во всем мире, еще реже встречается дерматоспараксис – описано 10 случаев в мире [5].
КЛИНИЧЕСКИЕ ПРИЗНАКИ И ДИАГНОСТИКА
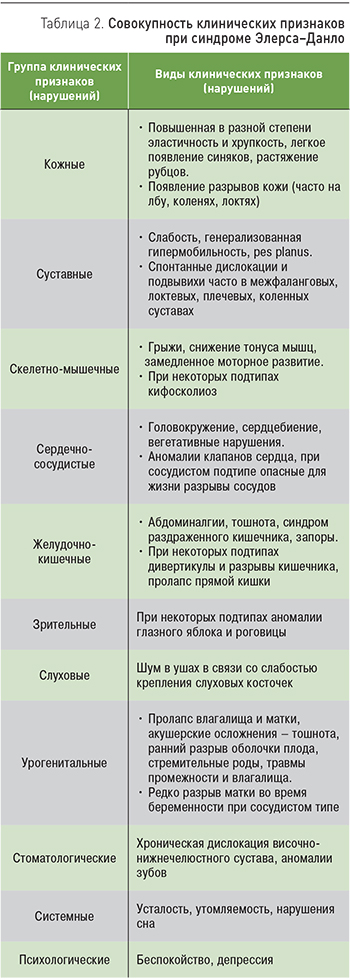
Для большинства подтипов СЭД характерными симптомами являются изменения кожи и суставов [4, 7]. Вместе с тем нарушения соединительной ткани присутствуют во многих органах и изменяют их функциональное состояние. В целом совокупность клинических признаков СЭД (табл. 2) зависит от подтипов заболевания [2, 6, 7].
Диагностика СЭД начинается с физикального обследования и включает:
- выявление клинических признаков, характерных для СЭД;
- оценку гипермобильности суставов с использованием шкалы Beighton;
- поиск аномальных рубцов и тестирование кожи (мягкость, хрупкость, растяжимость);
- выявление осложнений, обусловленных СЭД;
- изучение семейного анамнеза [5, 6].
Генетическая гетерогенность и фенотипическая изменчивость СЭД обусловливает клиническое сходство между подтипами заболевания, а также другими наследственными нарушениями соединительной ткани (ННСТ). Окончательный диагноз СЭД необходимо подтверждать молекулярным тестированием. Для большинства подтипов СЭД идентифицированы мутации генов, кодирующих коллаген, или генов, кодирующих коллагенообразующие ферменты. В случае отсутствия генетического тестирования проводят электронную микроскопию биоптатов кожи с оценкой структуры коллагена. Отсутствие результатов, подтверждающих предполагаемый диагноз, не исключает СЭД [5, 6].
 ОБЩИЕ ПРИНЦИПЫ ЛЕЧЕНИЯ
ОБЩИЕ ПРИНЦИПЫ ЛЕЧЕНИЯ
Несмотря на то что пациенты с мягкими проявлениями СЭД не нуждаются в специализированном лечении, необходимы консультации многих специалистов для оценки органных изменений. При тяжелых проявлениях синдрома лечение и наблюдение осуществляют профильные специалисты. Конкретные стратегии лечения зависят от типа СЭД.
Для пациентов всех типов рекомендуют:
- физиотерапевтические методы лечения (при мышечной гипотонии, осложнениях гипермобильности суставов и т.п.);
- психотерапию (при хронической боли, вегетативных расстройствах);
- симптоматическое лечение в зависимости от характера нарушений, целипролол (селективный β-адреноблокатор с внутренней симпатомиметической активностью) для предотвращения диссекции и разрыва артерий при сосудистом подтипе СЭД;
- бережное отношение к тканям и коже при травмах и оперативных вмешательствах;
- аскорбиновую кислоту при кровоточивости;
- генетическое консультирование [7].
ДИАГНОСТИКА ОТДЕЛЬНЫХ ПОДТИПОВ СЭД
Рассмотрим диагностические признаки наиболее распространенных подтипов СЭД.
1. Клинические критерии классического подтипа СЭД (в каталоге генов и генных болезней – OMIM#130000) отражены в табл. 3.
Окончательный диагноз классического подтипа СЭД требует подтверждения молекулярным тестированием. У большинства пациентов (>90%) с классическим типом СЭД диагностируют гетерозиготную мутацию в одном из генов, кодирующих коллаген типа V (COL5A1 и COL5A2), редко специфические мутации в генах, кодирующих коллаген типа I.
В случае отсутствия генетического тестирования диагностическое предположение подкрепляют результаты электронной микроскопии биоптатов кожи. Впрочем, отсутствие этих подтверждающих результатов не исключает диагноза, так как определенные типы мутаций (например, глубокие интронные мутации) могут не обнаруживаться стандартными диагностическими молекулярными методами. Вместе с тем альтернативные диагнозы следует рассматривать при отсутствии мутаций COL5A1, COL5A2, COL1A1 или COL1A2.
2. Сосудистый тип (OMIM #130050)
Это один из самых тяжелых типов СЭД, который имеет аутосомно-доминантное наследование с гетерозиготной мутацией генов, кодирующих коллаген типа III. Редко при сосудистом типе выявляют мутацию в гене COL1A1. Течение сосудистого типа СЭД нередко катастрофическое в связи с внезапным разрывом артерии. У беременных женщин риск смерти при его наличии составляет 5,3%.
Распространенность сосудистого типа СЭД 1:50 000–200 000 населения [12]. Исследователи считают, что мягкие фенотипические формы, вероятно, не диагностируют [6, 7].
Принципы диагностики этого типа СЭД отражены в табл. 4.
Клинический диагноз сосудистого подтипа вызывает трудности. Молекулярная диагностика основывается на гетерозиготной мутации в гене COL3A1, кодирующем коллаген типа III.
3. Гипермобильный тип (OMIM #130020)
Диагноз гипермобильного типа СЭД основывается на клинических данных, так как молекулярные генетические нарушения неизвестны, что, вероятно, указывает на гетерогенность типа. Его наследование аутосомно-доминантное. Поскольку лабораторное подтверждение/опровержение диагноза отсутствует, этот тип СЭД необходимо дифференцировать с другими синдромами ННСТ на основании клинических признаков. Диагностика гипермобильного типа СЭД возможна в случае выявления 3 характерных критериев [6, 13].
Критерий 1 – генерализованная гипермобильность суставов (ГМС) с оценкой в баллах по шкале Бейтона с учетом возраста пациента: ≥6/9 для детей и подростков, ≥5/9 для мужчин и женщин до 50 лет, ≥4/9 для мужчин и женщин старше 50 лет.
Для определения ГМС у пациентов пожилого возраста разработан опросник Хакима–Грэхема [14]. Положительный ответ на ≥2 вопросов предполагает генерализованную ГМС с чувствительностью 80–85% и специфичностью 80–90%.

Критерий 2 – не менее 2 следующих характеристик (А-С):
А – системное вовлечение соединительной ткани (≥ 5 признаков):
1. мягкая бархатистая текстура кожи;
2. легкая ГМС;
3. необъяснимые стрии на спине, бедрах, в паховой области, груди или на передней стенке живота у подростков, мужчин или нерожавших женщин без значительных колебаний веса;
4. двусторонние пьезогенные папулы на пятках;
5. рецидивирующие или множественные абдоминальные (пупочная, паховая) боли или боли в области голени грыжи;
6. атрофические рубцы, по меньшей мере на двух участках, без образования папиросных рубцов или участков гемосидероза;
7. пролабирование тазового дна, прямой кишки или матки у детей, мужчин или женщин без ожирения или других предшествующих состояний;
8. скученность зубов и высокое или узкое нёбо;
9. арахнодактилия, выявленная при положительном тесте Steinberg и тесте запястья;
10. отношение размах рук к росту ≥1,05;
11. пролапс митрального клапана умеренный или значительный по эхокардиографическим критериям;
12. дилатация корня аорты Z ≥2.
B – положительная семейная история с ≥1 родственником независимо от признаков СЭД в настоящее время;
C – костно-мышечные признаки (≥1 признака):
1. боль в области опорно-двигательного аппарата в двух или более конечностях, повторяющаяся ежедневно в течение ≥3 мес;
2. хроническая распространенная боль в течение ≥3 мес;
3. рецидивирующая дислокация суставов или нестабильность в отсутствие травм (а и б):
а) ≥3 нетравматических дислокаций в одном и том же суставе или 2 эпизода нетравматической дислокации в ≥2 разных суставах, возникающие в разное время;
б) медицинское подтверждение нестабильности в ≥2 суставах, не связанной с травматическим поражением.

Критерий 3:
1. отсутствие необычной хрупкости кожи, которая должна побуждать к рассмотрению других типов СЭД;
2. исключение других наследуемых или приобретенных нарушений соединительной ткани, в том числе аутоиммунных;
3. исключение альтернативных диагнозов, сопровождающихся гипермобильностью или мышечной гипотонией, другие ННСТ (нейромышечные заболевания, синдром Марфана), скелетные дисплазии.
Подтверждением других заболеваний и состояний служат анамнез, клинические данные и молекулярно-генетическое исследование.
Помимо основных критериев гипермобильного типа СЭД, встречаются симптомы, которые не обладают высокой чувствительностью и не включены в диагностические признаки: нарушение сна, усталость, постуральная тахикардия, функциональные желудочно-кишечные расстройства (абдоминалгия, синдром раздраженного кишечника), беспокойство, депрессия, панические атаки, дисфункция мочевого пузыря, склонность к тошноте, генерализованная боль, головная боль [18].
