КОЛЛЕКТИВ ЭКСПЕРТОВ:
Проф. Л.Б. Лазебник (Москва), д.м.н. Е.В. Голованова (Москва), проф. С.А. Алексеенко (Хабаровск), д.м.н. А.О. Буеверов (Москва), проф. Е.Ю. Плотникова (Кемерово), проф. А.И. Долгушина (Челябинск), д.м.н. Л.Ю. Ильченко (Москва), к.м.н. Т.В. Ермолова (Санкт-Петербург), д.м.н. Л.В. Тарасова (Чебоксары), д.м.н. Е.Д. Ли (Москва), Ю.В. Цыганова (Чебоксары), проф. В.А. Ахмедов (Омск), Е.А. Агеева (Хабаровск), к.м.н. В.М. Лосев (Самара), к.м.н. И.Н. Куприянова (Екатеринбург), д.м.н. С.Н. Серикова (Краснодар), проф. Н.В. Корочанская (Краснодар), к.м.н. Л.Г. Вологжанина (Пермь), д.м.н. Я.С. Циммерман (Пермь), д.м.н. Е.И. Сас (Санкт-Петербург), д.м.н. С.В. Журавель (Москва), д.м.н. С.В. Оковитый (Санкт-Петербург), проф. М.Ф. Осипенко (Новосибирск), д.м.н. В.Г. Радченко (Санкт-Петербург), проф. Г.С. Солдатова (Новосибирск), к.м.н. С.И. Ситкин (Санкт-Петербург), к.м.н. П.В. Селиверстов (Санкт-Петербург), проф. Г.В. Шавкута (Ростов-на-Дону), к.м.н. Е.Н. Бутова (Ростов-на-Дону), к.м.н. С.А. Кожевникова (Воронеж).
Человеческий организм, как и всякий другой, является промежуточным компонентом круговорота азота в природе. Потребляя азот из внешней среды в виде различных соединений, организм перерабатывает его в аммиак – один из конечных продуктов обмена азотсодержащих веществ [1], который выводится из организма в виде мочевины.
Наиболее активными продуцентами аммиака выступают органы с высоким обменом аминокислот и биогенных аминов – нервная ткань, печень, кишечник, мышцы.
В состоянии азотистого равновесия организм взрослого человека потребляет и выделяет около 15 г азота за сутки. К временному или постоянному нарушению азотистого баланса приводит огромное количество физиологических состояний и заболеваний, и необходимость его стабилизации хорошо известна. Вместе с тем, несмотря на огромное количество исследований, посвященных роли метаболизма азота и его соединений в клинике, до настоящего времени мы не смогли обнаружить в мировой литературе какой-либо согласительный документ по классификации уровня аммиака-аммония в крови человека и подходах к коррекции гипераммониемии, что и явилось основой для появления данного консенсуса.
ПОЛОЖЕНИЕ 1
Взаимно трансформирующиеся, биологически активные молекулы (газ аммиак NH3 и катион аммония NH4+) являются конечными продуктами катаболизма белка, вырабатываются собственными тканями и уреаза-продуцирующими бактериями организма.
Метаболизация аммиака происходит преимущественно в клетках печени и мышечной ткани путем синтеза мочевины и глутамина, которые выводятся из организма с мочой, калом и выдыхаемым воздухом.
Основные механизмы образования аммиака в организме человека:
- неокислительное дезаминирование некоторых аминокислот (серина, треонина, гистидина) в печени;
- окислительное дезаминирование глутаминовой кислоты во всех тканях (кроме мышечной), особенно в печени и почках;
- дезаминирование амидов глутаминовой и аспарагиновой кислот в печени и почках;
- катаболизм биогенных аминов во всех тканях, в наибольшей степени в нервной ткани;
- гидролитическое дезаминирование в интенсивно работающих мышцах;
- распад аминокислоты глутамина – основного источника энергии клеток слизистой оболочки кишечника в тонкой кишке;
- распад пуриновых и пиримидиновых оснований – во всех тканях;
- осуществление жизнедеятельности уреаза-продуцирующих микроорганизмов в желудке, толстой кишке и мочевыводящих путях.
Значительные количества аммиака образуются в результате метаболизма кишечных бактерий в толстой кишке, откуда он поступает в кровь воротной венозной системы. В нормальных условиях печень быстро извлекает аммиак из крови воротной вены и обезвреживает его, поэтому кровь, выходящая из печени, практически не содержит аммиака [2].
Аммиак – токсичный газ, у здорового человека он присутствует в крови в относительно небольших концентрациях (25–40 мкмоль/л). Содержание свободного аммиака в крови представлено лишь следовыми количествами: не более 1% вещества в водной среде крови циркулирует в свободной форме.
Нормальная концентрация аммиака в крови не определена международными стандартами, поскольку зависит от методики и реактивов, применяемых в лаборатории. При этом допустимый уровень аммиака в крови (нормоаммониемия) обычно не превышает 60 мкмоль/л [3, 4].
Симптомы хронической интоксикации наблюдаются при превышении содержания аммиака в 2–3 раза, но даже незначительное его повышение (на 30–50%) оказывает неблагоприятное воздействие на организм и прежде всего на центральную нервную систему, что проявляется в виде головной боли, быстрой утомляемости, сонливости и др. (бытовая усталость, профессиональное переутомление, нервное или физическое перенапряжение и т.д.).
Практически весь аммиак удаляется из организма через почки с мочой в виде мочевины, которая синтезируется в печени, и в виде солей иона аммония, образующихся в эпителии канальцев почек. В клетки печени и почек аммиак попадает в составе транспортных форм (глутамина, аспарагина, глутаминовой кислоты и аланина), которые представляют собой нетоксичные соединения, образующиеся в процессе обезвреживания (связывания) аммиака [5].
Синтез мочевины – основной путь обезвреживания аммиака. На долю мочевины приходится до 80–85 % от всего выводимого из организма азота.
Количество выделяемой мочевины зависит от количества белков, поступающих с пищей. Если суточный рацион содержит 80–100 г белка, то за сутки образуется и выводится 25–30 г мочевины. Процесс образования мочевины происходит в печени в «орнитиновом цикле» (цикл Кребса–Гензелейта), который выполняет 2 функции: а) превращение азота аминокислот в мочевину, что предотвращает накопление токсичного аммиака; б) синтез аргинина и пополнение его депо в организме. В цикле принимают участие две аминокислоты, которые не входят в состав белков – орнитин и цитруллин, и две протеиногенные аминокислоты – аргинин и аспарагиновая кислота. Процесс включает пять реакций: первые две протекают в митохондриях, остальные – в цитозоле гепатоцитов.
Для синтеза 1 моля мочевины, который происходит в печени в орнитиновом цикле, используется 1 моль аммиака и 1 моль аспарагиновой кислоты. В сутки для синтеза 25 г мочевины затрачивается 6,3 г аммиака и 50 г аспартата.
При снижении детоксикационнной функции печени нарушается превращение аммиака в мочевину, в результате чего активируются альтернативные метаболические пути превращения аммиака в мышечной системе, астроцитах центральной нервной системы и почках [6, 7, 8]. При этом основными органами детоксикации аммиака становятся скелетная мышечная (что сопровождается развитием саркопении) и нервная ткани (развиваются полиневриты и энцефалопатия) [9].
В частности, в астроцитах начинается процесс превращения глутамата в глутамин при активном вовлечении глутаминсинтетазы. При наличии стойкой гипераммониемии, кроме повышения активности глутаминсинтетазы, изменяется активность других ферментов, в частности белка – переносчика глутамата [10]. Это приводит к повышению концентрации глутамата в экстрацеллюлярном пространстве и активации процессов цитотоксичности, свойственной возбуждающим нейротрансмиттерам (эксайтотоксичность) [11]. Таким образом, ферменты глутаматдегидрогеназа и глутаминсинтетаза являются регуляторными и обусловливают скорость процессов образования и обезвреживания аммиака. Некоторые ферменты мочевинообразования присутствуют в головном мозге, эритроцитах, сердечной мышце, однако весь набор энзимов имеется только в печени.
Длительная гипераммониемия может сопровождаться образованием дефектных нейротрансмиттеров, что сопровождается изменением активности серотонин- и глутаматергической систем, с нарастанием активности ГАМК-ергической системы [12].
Кроме этого, стабильно повышенный уровень азотистых соединений приводит к избыточному образованию свободных радикалов, в результате чего происходит дальнейшая дестабилизация функционального состояния астроцитов. В головном мозге к гипераммониемии наиболее чувствителен гипоталамический центр [11], что проявляется стойкой анорексией и формированием белково-энергетической недостаточности, усугубляющей катаболические процессы.
Продуцируемый миоцитами миостатин не только мощно подавляет рост мышечной массы, но и приводит к ее уменьшению [13]; при гипераммониемии у больных с циррозом печени клинически это проявляется синдромом саркопении [13, 14, 15]. Гипераммониемия индуцирует аутофагию, когда поврежденные белки расщепляются, но не перерабатываются [16, 17].
Саркопения – один из прогностических факторов летального исхода при циррозе печени, что подтверждается низкими показателями шкалы MELD (Model for the End stage Liver Disease – оценка терминальных стадий заболеваний печени и прогноза доимплантационной выживаемости) [9, 14]. В исследованиях выявлена корреляция между дефицитом белков и риском формирования сепсиса [10].
ПОЛОЖЕНИЕ 2
Существуют различные методики определения показателей нормального содержания аммиака (NH3) и аммония (NH4+) в средах организма, среди которых распространенными являются иммуноферментный метод (ИФА) и экспресс-метод. Информативным и наиболее легко осуществляемым считается экспресс-метод фотометрического количественного определения аммиака в капиллярной крови с помощью портативного анализатора PocketChem.
При физиологическом рН 97–99% аммиака присутствует в крови в ионизированной форме – в виде аммония NH4+.
Прямой ферментативный метод определения уровня аммиака в крови (аммониемии) исторически используется наиболее часто и представляет собой реакцию, в которой аммоний в присутствии глутаматдегидрогеназы вступает во взаимодействие с α-оксоглутаратом и редуцирует содержание никотинамидадениндинуклеотида фосфата (NADPH) с образованием глутамата, NADP+ и воды. Уровень содержания аммиака варьирует в образцах венозной, капиллярной и артериальной крови, но в норме обычно находится в диапазоне от 11 до 50 мкМ [18, 19]. Хотя аммиак артериальной крови считается более достоверным показателем его системного содержания, обычно используется определение аммиака в венозной крови, уровень которого несколько отличается от артериального [20, 21].
Основное условие получения достоверных результатов измерения аммиака в крови – строгое соблюдение условий технологии правильного забора, обработки и доставки образца крови. Так, полученный для транспортировки в лабораторию материал следует хранить при температуре ниже 0 °C и отделить плазму от эритроцитов в течение 15 мин после сбора. Эти жесткие условия по времени доставки образца крови для исследования требуют пребывания пациента в непосредственной близости к клинической лаборатории. После отделения плазмы аммиак стабилен в течение 4 ч при температуре 4 °С.
Важно отметить, что сопровождающие забор крови травмирование сосуда и гемолиз эритроцитов могут приводить к спонтанному увеличению уровня аммиака [22, 23, 24]. Стресс, физические упражнения, психологическая усталость, табакокурение, нарушение питания и употребление алкоголя могут также влиять на уровень системного аммиака. Из-за высокого риска ложного результата рекомендуется повторить исследование в то же время суток и при тех же условиях, чтобы подтвердить результат.
Существуют также методы определения аммиака в эритроцитах, выдыхаемом воздухе, слюне, поте и моче [25].
Измерение уровня аммиака в выдыхаемом воздухе относится к неинвазивным, легко применяемым комфортным методам диагностики для всех групп пациентов (Hibbardand Killard, 2011). Никакой обработки выдыхаемого воздуха не требуется, что является преимуществом метода; в то же время не получено убедительных доказательств корреляций уровней аммиака в выдыхаемом воздухе и артериальной крови [26, 27, 28].
Определение уровня аммиака в ткани печени может быть использовано для пациентов, нуждающихся в диагностической биопсии печени. Следует учитывать, что колориметрические наборы, используемые для определения аммиака в образцах тканей, довольно дороги и чувствительны к другим источникам аммиака, присутствующим в окружающей среде, что требует работы в специальной установке или зоне отрицательного воздушного давления, которые не всегда доступны для всех лабораторий. Более того, длительное хранение ткани даже при температуре -80 °C может влиять на стабильность образцов, что приводит к искажению результата ниже ожидаемого.
В 2018 г. в России зарегистрирован портативный экспресс-анализатор количественного определения аммиака в крови – PocketChem-4140. Соответствующий метод определения – фотометрический, основанный на микродиффузии, т.е. косвенный. Время определения концентрации аммиака составляет около 200 с.
Для исследования забирается капиллярная кровь из пальца, однако технология забора требует соблюдения определенных условий, изложенных в инструкции. Используются только свежие образцы крови. В приборе образовавшийся из аммония аммиак проходит через полупроницаемую мембрану и изменяет цвет индикатора, длина волны которого подвергается спектроскопическому анализу и автоматически указывает количественное его содержание. Калибровка прибора и коррекция результатов осуществляются автоматически.
ПОЛОЖЕНИЕ 3. Классификация гипераммониемий
 Сводная классификация гипераммониемий представлена в таблице 1.
Сводная классификация гипераммониемий представлена в таблице 1.
В традиционной клинической практике выделяют два основных варианта гипераммониемий [29]:
A. Наследственные (врожденные) гипераммониемии – результат различных генетических дефектов ферментов цикла образования мочевины;
B. Приобретенные гипераммониемии:
a) печеночные – следствие снижения активности орнитинового цикла и глутаминсинтетазной реакции за счет печеночно-клеточной недостаточности, портосистемного шунтирования крови, развития и прогрессирования портальной гипертензии;
b) внепеченочные – следствие нарушения метаболизма аммиака, не связанного с патологией печени.
К настоящему времени описаны многочисленные причины внепеченочных (нецирротических) гипераммониемий [30, 31]:
I. Состояния, связанные с увеличением производства аммиака:
- инфекции уреаза-продуцентами: Proteus mirablis, Klebsiella species, Escherichia coli, Morganella morganii, Providencia rettgeri, Corinobacteria (возбудители дифтерии), Mycobacterium genavense, Herpes simplex [32], а также, вероятно, Helicobacter pylori (в частности, имеются сообщения о большей частоте встречаемости неалкогольной жировой болезнью печени у больных с хеликобактериозом [32, 33]);
- гематоонкологические расстройства: множественная миелома, химиотерапия острого лейкоза, трансплантация костного мозга, применение 5-фторурацила [34];
- трансплантация органов [34];
- нагрузка белками и увеличение катаболизма: тяжелые физические упражнения, судороги, голод или травма, общее парентеральное питание, желудочно-кишечные кровотечения, использование стероидов, бариатрическая хирургия.
Описаны случаи постбариатрической гипераммониемической энцефалопатии после хирургического шунтирования желудка по Roux-en-Y (RYGB) – самой распространенной процедуры снижения веса, проводимой в США [35, 36, 37]. Высокий риск гипераммониемической энцефалопатии после операции Roux-en-Y, при которой смертность приближается к 50%, установлен у женщин в возрастной группе 34–69 лет, даже не имевших в анамнезе какого-либо заболевания печени [36–48]. Угроза фатального исхода может быть обусловлена и тем фактом, что постбариатрическая гипераммониемия не сопровождается какими-либо изменениями биохимических показателей печеночного метаболизма. Гипераммониемия может наблюдаться у женщин в период беременности. развивается при нарушении гидролитического дезаминирования в интенсивно работающих мышцах, при эклампсии и в родах. У взрослых частичный дефицит ферментов может проявляться во время стрессовых заболеваний, таких как послеродовый стресс, острая кишечная инфекция, синдром короткой кишки, парентеральное питание с высоким потреблением азота, трансплантация сердца и легких и желудочно-кишечные кровотечения [39, 49, 50].
II. Состояния, связанные со снижением выделения аммиака:
- уретросигмоидостомия;
- портосистемные шунты, врожденная внутрипеченочная и внепеченочная гипертензия;
- применение некоторых лекарственных препаратов: вальпроевой кислоты, глицина, карбамазепина, рибавирина, сульфадиазина, пириметамина, салицилатов;
- врожденные дефекты метаболизма: нарушения орнитинового цикла, дефекты β-окисления жирных кислот и органических кислот, нарушение метаболизма пирувата.
ПОЛОЖЕНИЕ 4
Физиологические (функциональные) гипераммониемии включают следующие виды: постпрандиальная (на фоне высокобелковой диеты), после физических нагрузок (спортивная), после психогенных перегрузок (постстрессорная), вызванные прочими причинами (например, беременность).
Физиологическая гипераммониемия является признаком метаболических нарушений в мышечной ткани и часто проявляется состоянием утомления, что обусловлено нарушением ресинтеза АТФ. Повышение активности аденилатциклазного механизма при нарушении ресинтеза АТФ увеличивает образование ионов аммония, сдвигая метаболизм в сторону избыточного образования лактата, развития ацидоза, усиленной вентиляции легких и гиперпноэ [51, 52].
Высокобелковая диета, голодание, переедание, интенсивные физические нагрузки (в основном у мужчин и культуристов), тотальное парентеральное питание, роды также приводят к увеличению аммония в организме.
Во время анаэробных физических упражнений аммиак метаболизируется до мочевины в гепатоцитах и скелетных мышцах, мышечная усталость возникает в результате физических упражнений и приводит к заметному снижению производительности. Истощение мышечных сокращений, накопление продуктов метаболизма энергетических ресурсов, дисбаланс внутренней среды выделяются как основные факторы физической и психологической усталости. Содержание аммиака значительно повышается в случаях высокобелковой диеты после голодания [53].
ПОЛОЖЕНИЕ 5
Врожденные гипераммониемии обусловлены дефицитом ферментов орнитинового цикла – карбамоил-фосфат-синтетазы (гипераммониемия I типа) и орнитин-карбамоил-трансферазы (гипераммониемия II типа) и наблюдаются, как правило, в раннем детском возрасте.
В зависимости от дефицита или дефекта того или иного фермента выделяют несколько видов генетических заболеваний:
- гипераммониемию типа I (в основе – дефект карбамоилфосфатсинтетазы I);
- гипераммониемию типа II (в основе – дефект орнитинкарбамоилтрансферазы);
- цитруллинемию (в основе – дефект аргининосукцинатсинтетазы);
- аргининосукцинатурию (в основе – дефект аргининосукцинатлиазы);
- гипераргининемию (в основе – дефицит аргиназы).
ПОЛОЖЕНИЕ 6
По патогенезу приобретенные гипераммониемии следует разделять на:
- гепатогенные;
- гастроэнтерогенные;
- урогенные;
- сосудистые (портальная гипертензия);
- послеоперационные;
- прочие (лекарственные, в том числе цитостатические, лучевая терапия, онкологические, онкогематологические).
Приобретенная гипераммониемия может иметь внепеченочное происхождение [31, 51, 54]. Токсическое превышение аммиака регистрируется при гиповолемии, кровотечении из различных отделов желудочно-кишечного тракта (ЖКТ) у пациентов без цирроза печени, а также при сердечной недостаточности, легочном сердце, шунтирующих операциях, некоторых эндокринных нарушениях (декомпенсированном сахарном диабете, тяжелом тиреотоксикозе) и др. [52].
Гипераммониемическая энцефалопатия относится к редким осложнениям множественной миеломы, резистентной к химиотерапии [55]. Ее возникновение описано при лейкозе вследствие катаболических процессов [34], а также при приобретенном дефиците ферментов орнитинового цикла синтеза мочевины на фоне микровезикулярного ожирения печени (синдром Рейе), нарушении перфузии печени, метаболическом алкалозе и ацидозе, синдроме избыточного бактериального роста, длительных запорах.
Гипераммониемия может наблюдаться при любых патологических состояниях, сопровождающихся повышенным катаболизмом белков (массивных кровопотерях, обширных ожогах, синдроме сдавления или размозжения тканей, обширных гнойно-некротических процессах, гангрене конечностей, гипертермии различного происхождения, сепсисе и т.д.). Эти нарушения вызывают истощение мышечной массы тела, антиоксидантной защиты, а также значительно ослабляют и подавляют иммунитет. Повышение аммиака, нарушение баланса между ионизированной и неионизированной формой регистрируют при аутизме, не исключается его роль в развитии болезни Альцгеймера. Установлено, что вирусы острых респираторных инфекций приводят к снижению активности основного фермента орнитинового цикла – карбамоилфосфатсинтетазы; вследствие этого происходит накопление в крови субстрата этого фермента и его предшественников.
Гипераммониемия может наблюдаться при наличии аномалий нижних отделов мочевыводящей системы, которые вызывают затруднение оттока мочи, осложняющееся присоединением инфекции, обусловленной уреаза-продуцирующими бактериями (Proteusspecies, Corynebacteriumspecies, Klebsiellaspecies, Morganella morganii и др.). При этом образующийся свободный аммиак диффундирует в кровь [56].
Наряду с экзогенной причиной гипераммониемии, обусловленной протеиновой нагрузкой, описана генетическая непереносимость белка. Так, лизинурическая непереносимость протеинов, которую вызывают мутации в гене SLC7A7, определяющем мембранный транспорт основных аминокислот (орнитина, аргинина и лизина), приводит к дефициту и нарушению их всасывания в ЖКТ и реабсорбции в почках. Уровень аммиака при отказе от белковой пищи соответствовал нормальным показателям, но значительно повышался после приема белковой пищи, приводя к развитию комы [57].
Повышению содержания аммиака в крови способствует прием ряда лекарственных препаратов, а именно салицилатов, тетрациклина, глюкокортикостероидов, аспарагиназы, 6-азауридина, аллопуринола, тиазидных диуретиков, этакриновой кислоты, изониазида, карбамазепина и др. [58].
Вальпроат-индуцированная гипераммониемическая энцефалопатия у пациентов с эпилепсией является редким, но иногда смертельным осложнением. Повышение нейротоксина в этих случаях обусловлено истощением N-карбамилглутамата и может развиться как остро, так и при длительном приеме препарата [59]. Механизм лекарственно-обусловленной гипераммониемии не всегда ясен. Среди рисков ее развития следует учитывать присоединение инфекции, гиповолемию, наличие запоров и др.
Гипераммониемия также может развиться вследствие употребления большого количества алкоголя и психоактивных наркотических веществ.
Выкуривание одной сигареты приводит к повышению уровня аммиака в крови на 10 мкмоль/л.
ПОЛОЖЕНИЕ 7
Наиболее изученные клинические проявления гипераммониемии:
- печеночная энцефалопатия (в том числе минимальная и «скрытая, латентная», без видимых клинических проявлений);
- субклиническая печеночная недостаточность за счет эндотелиальной дисфункции;
- возможная активация звездчатых клеток печени, ведущая к печеночному фиброгенезу.
Скрининговым методом выявления энцефалопатии может служить тест связи чисел (ТСЧ).
При патологии печени наиболее частым проявлением гипераммониемии выступает печеночная энцефалопатия (ПЭ).
Существует несколько теорий и этапов патогенеза печеночной энцефалопатии [6, 7, 29]:
- связывание аммиака при синтезе глутамата вызывает отток α-кетоглутарата из цикла трикарбоновых кислот, при этом понижается образование энергии АТФ и ухудшается деятельность клеток;
- ионы аммония NH4+ вызывают защелачивание плазмы крови, при этом повышается сродство гемоглобина к кислороду (эффект Бора), гемоглобин не отдает кислород в капиллярах, в результате чего наступает гипоксия клеток;
- накопление свободного иона NH4+ в цитозоле влияет на мембранный потенциал и работу внутриклеточных ферментов, поскольку он конкурирует с ионными насосами для Na+ и K+;
- продукт связывания аммиака с глутаминовой кислотой – глутамин – является осмотически активным веществом. Его накопление приводит к задержке воды в клетках и развитию отека тканей. В частности, накопление его в нервной ткани может вызвать отек мозга, кому и смерть;
- глутамин удаляется из ЦНС посредством транспортной системы, которая функционирует путем обмена, поэтому увеличение скорости выхода аммиака также сопровождается нарастанием транспорта из крови ароматических аминокислот, которые вызывают торможение ферментативной системы, в норме превращающей тирозин в дофамин и норадреналин. В результате метаболизм исходных соединений протекает с образованием ложных нейротрансмиттеров, сходных по структуре с истинными адренергическими нейротрансмиттерами, которые замещают последние на уровне нервно-мышечных синапсов, но с эффективностью проведения нервного импульса меньше в 50 раз;
- накопление в ЦНС ложных нейротрансмиттеров (октопамин, фенилэтиламин, тирамин, фенилэтиноламин), а также продукта метаболизма триптофана – серотонина, являющегося нейротрансмиттером с преимущественно ингибиторным эффектом, способствует угнетению нервной системы, истощению функций мозга и развитию энцефалопатии;
- накопление аммиака и меркаптанов в крови вызывает быстрое прогрессирование печеночной недостаточности вследствие их выраженного угнетающего действия на регенерацию гепатоцитов.
Таким образом, в основе патогенеза ПЭ лежит дисбаланс аминокислот в головном мозге, приводящий к отеку астроглии и нарушениям ее функций, таких как изменения постсинаптических рецепторов и процессов нейротрансмиссии, нарушение проницаемости гематоэнцефалического барьера (ГЭБ), снижение энергетического обеспечения нейронов. Степень клинических проявлений нередко коррелирует с уровнем аммиака в сыворотке крови, в зависимости от выраженности нарушений деятельности головного мозга выделяют четыре степени тяжести печеночной энцефалопатии (табл. 2).
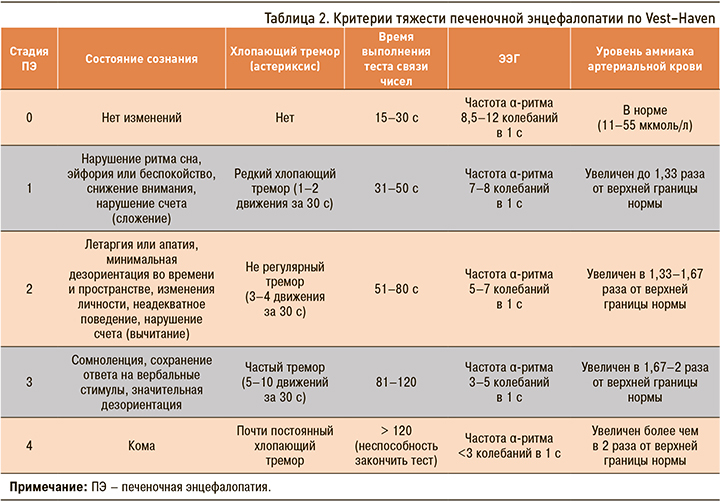
В последние годы в связи с совершенствованием диагностических методик актуально отделение клинически выраженных стадий ПЭ (дезориентация, атаксия, кома) от стадии с минимально выраженными проявлениями (латентной ПЭ).
Латентная ПЭ представляет наибольшую сложность для дифференциального диагноза, так как, несмотря на удовлетворительную ориентированность во времени и пространстве, наблюдаются изменения психометрических или нейропсихологических тестов. Имеющиеся при латентной ПЭ когнитивные и психомоторные расстройства (трудности с принятием решений, снижение скорости психомоторных реакций) нередко не воспринимаются больными и их окружением как проявление болезни.
Между тем результатом таких нарушений могут стать серьезные дорожно-транспортные происшествия с тяжелыми последствиями [60, 61]. В связи с этим выявление минимальной ПЭ имеет большое значение у работников многих профессий: водителей автотранспорта, операторов на автоматизированном оборудовании и др.
При клиническом осмотре на этой стадии могут выявляться некоторые нарушения когнитивных функций и/или поведения: некоторая дезориентированность, эйфория или тревога, снижение концентрации внимания, сложность выполнения операций по арифметическому сложению или вычитанию, изменение ритма сна [29].
Чаще всего латентную ПЭ можно выявить, только используя специальные опросники или метод вызванных потенциалов головного мозга. На данной стадии ПЭ необходимо дифференцировать ее с рядом состояний, сопровождающихся когнитивными нарушениями (табл. 3).

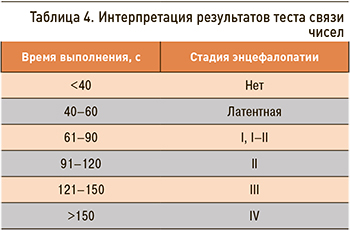 В связи с тем что имеется корреляция между уровнем гипераммониемии и стадией ПЭ, которую можно определить по ТСЧ, представляется возможным косвенное выявление наличия и выраженности гипераммониемии с использованием того же теста связи чисел (рис. 1). Степень выраженности энцефалопатии рассчитывается по скорости выполнения ТСЧ (табл. 4).
В связи с тем что имеется корреляция между уровнем гипераммониемии и стадией ПЭ, которую можно определить по ТСЧ, представляется возможным косвенное выявление наличия и выраженности гипераммониемии с использованием того же теста связи чисел (рис. 1). Степень выраженности энцефалопатии рассчитывается по скорости выполнения ТСЧ (табл. 4).
 Для удобства врача и пациента ТСЧ можно провести онлайн по адресу: www.Hepa-Merz.ru.
Для удобства врача и пациента ТСЧ можно провести онлайн по адресу: www.Hepa-Merz.ru.
ПОЛОЖЕНИЕ 8
Персистирующая и прогрессирующая гипераммониемия при хронических заболеваниях печени (неалкогольной жировой болезни печени) оказывает профиброгенный эффект, способствует нарушению печеночной гемодинамики, развитию портальной гипертензии, а в последующем саркопении скелетной мускулатуры.
В британском исследовании [68] in vitro и in vivo показано, что гипераммониемия служит причиной:
- активации звездчатых клеток печени;
- снижения клеточного метаболизма и пролиферации звездчатых клеток печени;
- активации профиброгенного/провоспалительного профиля звездчатых клеток;
- нарушения внутрипеченочной гемодинамики и нарастания портального давления;
- стимуляции эндоретикулярного стресса;
- индукции образования активных форм кислорода.
В экспериментальных и клинических исследованиях показано, что при неалкогольной жировой болезни печени (НАЖБП) гипераммониемия развивается на доцирротической стадии за счет снижения активности ферментов, участвующих в орнитиновом цикле [69–74]. У мышей с НАЖБП, развившейся на фоне жировой диеты, зафиксировано снижение экспрессии гена и белка карбамоилфосфатсинтетазы (CPS) и частично орнитинкарбамоилтрансферазы, что сопровождалось снижением синтеза мочевины и увеличением количества аммиака в ткани печени в сравнении с контрольной группой. У больных с НАЖБП на фоне ожирения выявлено достоверное, по сравнению со здоровыми лицами, снижение количества и активности ферментов оринитинового цикла, в частности, более чем в два раза снижен уровень орнитинтранскарбамилазы, причем на стадии стеатогепатита эти изменения выражены значительно сильнее (p <0,05), чем на стадии стеатоза [75, 76, 77].
В отечественном исследовании у больных хроническими заболеваниями печени (ХЗП) c фиброзом 0–1 стадии методом полигепатографии выявлены нарушения портопеченочной гемодинамики. У больных с фиброзом 0 стадии эти изменения носили динамический характер, что выявлялось на фоне функциональных тестов с глубоким дыханием и нитратами. Полученные данные свидетельствуют о том, что уже на ранних стадиях ХЗП формируются нарушения внутрипеченочной микроциркуляции различной локализации.
Также у больных ХЗП выявлены признаки дисфункции эндотелия: нарушение эндотелий-зависимой вазодилатации, повышение метаболитов оксида азота в периферической крови, нарушение экспрессии синтаз оксида азота в ткани печени (снижение экспрессии эндотелиальной синтазы и появление индуцибельной синтазы оксида азота) [78, 79].
ПОЛОЖЕНИЕ 9
Физиологическая (функциональная) гипераммониемия не нуждается в лечении.
Патогенетическая терапия гипераммониемии, вне зависимости от степени ее тяжести, направлена на лечение основного заболевания, включает диету с ограничением животного и достаточным количеством растительного белка, ограничение физических нагрузок, по показаниям назначение кишечных невсасывающихся антибиотиков (рифаксимин-альфа), пре- и пробиотиков.
L-орнитин-L-аспартат (LOLA) является базисным препаратом, позволяющим в качестве моно- или политерапии корректировать уровень гипераммониемии.
Современные методы лечения гипераммониемии нацелены на уменьшение аммониогенеза, абсорбцию аммиака в ЖКТ, активацию удаления аммиака путем активирования уреагенеза путем лечения основного заболевания или добавлением промежуточных продуктов цикла мочевины и синтеза глутамина [80].
Последние данные показывают, что чрезмерное ограничение белка может увеличить уровень аммиака в сыворотке крови в результате активации мышечного катаболизма. Кроме того, ограничение потребления белка ухудшает нутритивный статус, что сказывается отрицательно на самочувствии пациентов с печеночной энцефалопатией [81]. У пациентов с установленным диагнозом цирроза печени минимальный ежедневный диетический уровень потребления белка, необходимый для поддержания азотистого баланса, составляет 0,8–1,0 г/кг веса [82]. Поэтому в настоящее время ряд документов рекомендует при ПЭ нормопротеиновую диету, а больным, которые не переносят нормальный растительный белок, рекомендуется замена его животным белком [83–86].
L-орнитин L-аспартат (LOLA) – стабильная соль орнитина и аспарагиновой кислоты, которая представляет собой важный субстрат синтеза для глутамина и мочевины, основных компонентов дезаминирования [87]. Основные фармакологические свойства LOLA обусловлены участием обеих аминокислот, входящих в состав препарата, в орнитиновом цикле.
L-орнитин:
- включается в цикл мочевины в качестве субстрата (на этапе синтеза цитруллина);
- является стимулятором карбамоилфосфатсинтетазы I (первого фермента цикла мочевины);
- является активатором глутаминсинтетазной реакции в печени и мышцах, снижает концентрацию аммиака в плазме крови;
- способствует нормализации кислотно-основного равновесия организма;
- способствует продукции инсулина и соматотропного гормона;
- улучшает белковый обмен при заболеваниях, требующих парентерального питания.
L-аспартат:
- включается в цикл мочевины на этапе синтеза аргининсукцината;
- является субстратом для синтеза глутамина;
- участвует в связывании аммиака в перивенозной крови, гепатоцитах, мозге, других тканях;
- стимулирует синтез глутамина в мышцах и перивенозных гепатоцитах;
- оказывает стимулирующее действие на неактивные или пораженные клетки печени;
- стимулирует регенерацию, улучшает энергетические процессы в поврежденной ткани печени;
- участвует в цикле трикарбоновых кислот;
- обладает способностью проникать через мембраны клеток путем активного транспорта;
- внутри клетки участвует в процессах энергетического обмена, проходящих в митохондриях, за счет чего повышает энергетическое обеспечение ткани;
- обладает анаболическим действием на мышцы.
Таким образом, LOLA повышает толерантность к белку и обладает анаболическим действием, увеличивает энергетический потенциал клеток, усиливает утилизацию молочной кислоты. Мембраностабилизирующий эффект обусловливает антиоксидантное действие L-орнитин-L-аспартата, этот эффект особо значим при хронических заболеваниях печени, в первую очередь алкогольной этиологии. Кроме этого, аспартат встраивается в цикл Кребса, т.е. увеличивает синтез макроэргов и снижает образование молочной кислоты, что, в свою очередь, уменьшает проницаемость ГЭБ для токсических веществ.
Механизм действия L-орнитина-L-аспартата указывает на целесообразность включения этого препарата в схему лечения больных как с печеночной недостаточностью (особенно осложненной печеночной энцефалопатией), так и доцирротической и нецирротической гипераммониемией на самых ранних стадиях патологии. Длительность лечения зависит от степени выраженности гипераммониемии и может продолжаться в течение продолжительного времени.
Прием LOLA предотвращает дорожно-транспортные нарушения у водителей с хроническим гепатитом С и минимальным фиброзом печени [61].
Demura S. et al. в двойном слепом плацебо-контролируемом исследовании изучали влияние L-орнитина на переносимость велотренировок, скорость истощаемости и метаболизм аммиака во время и после тренировки у здоровых волонтеров. Концентрация аммиака в плазме сразу после и через 15 мин после дополнительных нагрузок в группе L-орнитина была значительно ниже, чем в группе плацебо. Таким образом, L-орнитин увеличивал способность аммиачного буфера как во время, так и после тренировки [84].
Гепатопротективные свойства LOLA показаны в отношении пациентов с ХЗП различной этиологии [88–91]. Данные многоцентрового нерандомизированного проспективного когортного исследования (1167 пациентов с ХЗП, в том числе 648 больных НАЖБП на стадии стеатогепатита) продемонстрировали эффективность и хорошую переносимость LOLA [91].
В исследованиях показано, что нарастание концентрации аммиака выявляется у больных с ХЗП уже на доцирротической стадии [88]. При этом у больных НАЖБП с 0–1 стадией фиброза на фоне применения L-орнитин-L-аспартата исходно имевшаяся гипераммониемия существенно снизилась и сопровождалась улучшением общего состояния и лабораторных показателей.
В другом российском исследовании с использованием реогепатографии было убедительно показано нарушение портального кровотока при гипераммониемии у больных с доцирротическими стадиями ХЗП [78, 79]. Лечение 289 пациентов с неалкогольным стеатогепатитом (НАСГ) с использованием LOLA на протяжении 3 мес на фоне хорошей переносимости и высокого комплаенса больных способствовало снижению уровней аммиака, коррелирующим с уменьшением сосудистых нарушений, статистически значимому улучшению клинико-биохимических показателей и повышению качества жизни [79]. L-орнитин-L-аспартат улучшал показатели внутрипеченочного кровотока у пациентов с различными типами нарушений портопеченочной гемодинамики. Улучшение печеночного кровотока может быть обусловлено в том числе деактивацией звездчатых клеток печени и снижения их контрактильности.
Основная цель назначения антибиотиков больным с гипераммониемией и ПЭ – подавление уреаза-продуцирующей кишечной микрофлоры [92]. Предпочтение отдается невсасывающемуся антибиотику широкого спектра действия рифаксимину-альфа. Он активен в отношении большинства как грамположительных, так и грамотрицательных, как аэробных, так и анаэробных бактерий. Препарат практически не всасывается в ЖКТ, при пероральном приеме натощак в крови обнаруживается не более 1% от принятой дозы. Минимальное всасывание действующего вещества в плазму крови снижает риск возникновения системных побочных эффектов, внекишечных лекарственных взаимодействий с другими препаратами, поэтому у пациентов с заболеваниями печени нет необходимости в коррекции дозы.
Положительное влияние рифаксимина-альфа на лечение гипераммониемии показано в нескольких клинических исследованиях [93, 94]. Эффективность применения этого лекарственного средства при лечении ПЭ была более высокой, чем при использовании невсасывающихся дисахаридов. Исследования, выполненные в соответствии с Good Clinical Practice (Надлежащая клиническая практика, ГОСТР 52379-2005), подтверждают, что рифаксимин-альфа может быть эффективнее лактулозы у ряда пациентов с ПЭ I–III стадий [92–98]. Рекомендуемая взрослым ежедневная доза 1200 мг/сут, как правило, разделяется на три приема, длительность лечения составляет 7–10 сут, повторение таких курсов рекомендуется проводить ежемесячно на протяжении длительного времени, при необходимости постоянно [93–94]. В ряде исследований показано уменьшение числа госпитализаций на фоне приема рифаксимина-альфа у пациентов с рецидивирующей энцефалопатией [99–102].
Применение пробиотиков позволяет конкурентно вытеснять уреаза-продуцирующие патогенные бактерии в кишечнике. Результаты исследований по оценке влияния пробиотиков на ПЭ [103, 104] показали, что они уменьшают проницаемость кишечника и секрецию бактериальной уреазы, увеличивают выделение аммиака, улучшают метаболический потенциал эпителия кишечника, играют роль в снижении концентрации аммиака в портальной крови, поскольку ингибируют бактериальную уреазную активность.
Поскольку большинство пробиотиков продуцирует кислоты, которые снижают рН в кишечнике, абсорбция аммиака уменьшается [105]. Кроме того, пробиотики способствуют уменьшению воспаления и окислительного стресса в клетках печени, что приводит к увеличению печеночного клиренса аммиака и уменьшению поглощения других токсинов.
В метаанализе 21 рандомизированного клинического исследования с участием 1420 пациентов с ПЭ показано, что пробиотики уменьшают клинические проявления ПЭ, повышают качество жизни больных и способствуют снижению концентрацию аммиака в плазме, но не влияют на показатель смертности [106]. Пероральный прием штаммов пробиотиков вида Lactobacillus снижает уровень аммиака в крови у пациентов с циррозом печени [107]. Штамм бактерий Lactobacillus acidophilus модифицирует кишечную флору, улучшает когнитивные функции у пациентов с циррозом печени, а штамм Enterococcus faecium SF68 повышает толерантность к белковой нагрузке, способствует достижению более низкого уровня аммиака, а также улучшает психическое состояние и психометрические показатели при длительном лечении пациентов с циррозом печени и ПЭ 1–2 степени [108, 109].
Высококонцентрированные комбинации пробиотических штаммов (Bifidobacterium sp., Lactobacillu ssp., Streptococcus thermophilus) у пациентов с циррозом и ПЭ оказывают существенное влияние на степень гипераммониемии [110].
В настоящее время пробиотики остаются второй или третьей линией терапии гипераммониемии и ПЭ. В то же время пребиотики лактитол и лактулоза при лечении печеночной энцефалопатии в ряде ситуаций могут быть средством выбора [111, 112].
Эксперты отмечают, что, помимо заболеваний печени, пока не получено доказанных корреляций между уровнем гипераммониемии и клинической картиной заболевания. Модели энцефалопатии и фиброза печени являются наиболее изученными и требуют вдумчивого и дифференцированного подхода к избранным способам лечения. Выявление причин повышения конечных продуктов метаболизма азота должно способствовать коррекции патологического процесса через воздействие на все звенья патогенеза.
