ВВЕДЕНИЕ
Кисты печени все чаще обнаруживаются как случайная находка при визуализации органов брюшной полости. Они могут быть простыми и единичными или множественными и ассоциированными с поликистозом других органов. Данная патология может наблюдаться при различных состояниях, среди которых врожденный поликистоз печени и почек, аутосомно-доминантная поликистозная болезнь почек, аутосомно-доминантный поликистоз печени, поликистоз печени, ассоциированный с аутосомно-доминантным поликистозом почек, аутосомно-рецессивный поликистоз почек с врожденным фиброзом печени.
Поликистозная болезнь печени (ПБП) – редкое наследственное заболевание, которое может развиваться как самостоятельное, изолированное состояние или же сочетаться с аутосомно-доминантной или аутосомно-рецессивной поликистозной болезнью почек со сложным механизмом развития. ПБП в настоящее время не имеет единого диагностического стандарта: диагноз обычно ставится, когда количество кист печени превышает 20 [1].
Изолированная аутосомно-доминантная ПБП имеет распространенность в общей популяции от 1 до 10 случаев на 1 млн человек, в то время как частота аутосомно-доминантной поликистозной болезни почек колеблется в пределах от 1 на 400 до 1 на 1000. При этом, по статистике, встречаемость поликистоза почек достигает 80–90% у пациентов с ПБП [2].
С развитием ПБП ассоциированы мутации в различных генах. На сегодняшний день выделяют 12 генов, ответственных за это заболевание. Тем не менее многочисленные случаи ПБП до сих пор генетически не изучены [3].
В предлагаемой статье приведены два клинических случая пациенток с ассоциированным поликистозом печени и почек.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ № 1
Пациентка К., 81 год, в июне 2021 г. обратилась в приемное отделение ГАУЗ «Республиканская клиническая больница (РКБ) Минздрава Республики Татарстан» с жалобами на разлитые боли по всему животу. Там же была она осмотрена хирургом, где ей был выставлен предварительный диагноз «Асцит неясного генеза. Анемия средней степени тяжести. Хроническая болезнь почек». Для дообследования пациентка была госпитализирована в гастроэнтерологическое отделение больницы.
Данные анамнеза: впервые симптомы проявились у пациентки в 2016 г., когда она начала отмечать увеличение объема живота, дискомфорт в правом подреберье. При обследовании был выявлен поликистоз печени и почек. Периодически пациентка получала стационарное лечение по месту жительства: диуретики (спиронолактон и фуросемид) с положительным эффектом. С апреля 2021 г. ее состояние ухудшилось: наблюдались нарастающая общая слабость и отечно-асцитический синдром.
Данные объективного осмотра. При поступлении в отделение РКБ состояние пациентки было расценено как средней степени тяжести: ясное сознание, иктеричность кожи и склер, увеличение живота за счет асцита, живот при пальпации мягкий, безболезненный (пальпация других органов брюшной полости была затруднительная из-за асцита), варикозно расширенные вены передней брюшной стенки, симметричные периферические отеки нижних конечностей. Артериальное давление (АД), пульс, диурез в норме, стул 1 раз в 2 дня. В отделении пациентке был выставлен предварительный диагноз «Асцит неясной этиологии. Цирроз печени? Канцероматоз брюшины? Поликистоз печени и почек. Мочекаменная болезнь, конкремент чашечки нижней трети левой почки. Хроническая болезнь почек, стадия 4 (СКФ 28 мл/ мин./1,73). Гипертоническая болезнь 3 стадии, риск 4. Хроническая сердечная недостаточность 2а. ФК 3. Анемия средней степени гипохромная».
Было проведено комплексное обследование пациентки для уточнения диагноза и определения дальнейшей тактики ее ведения.
Данные общего анализа крови: гипохромная анемия (гемоглобин – 78 г/л; эритроциты – 2,92×1012/л; гематокрит – 20,25%).
Данные биохимического исследования крови: повышение функциональных проб печени (ФПП) (билирубин общий – 66,0 мкмоль/л; билирубин связанный – 55,2 мкмоль/л; АЛТ – 55,2 Ед/л; АСТ – 33,0 Ед/л; ЩФ – 157,0 Ед/л, ГГТП – 71,0 Ед/л); азотемия (мочевина – 14,3 ммоль/л; креатинин – 148 мкмоль/л); гиперурикемия (547,0 мкмоль). Были выявлены признаки печеночно-клеточной недостаточности: гипоальбуминемия (45,8%), протромбиновое время 17,3 с, МНО 1,47, АЧТВ 46,1 с, снижение протромбина по Квику (51,4%). Вирусные маркеры гепатита и ВИЧ отрицательные. Дополнительно для исключения онкопатологии были исследованы уровни онкомаркеров СА 19-9, альфафетопротеина, ракового эмбрионального антигена (показатели в норме). При исследовании асцитической жидкости был выявлен экссудат (проба Ривальта положительная, обнаружены в большом количестве эритроциты, белок и уробилин, прозрачность мутная).
При ультразвуковом исследовании органов брюшной полости (УЗИ ОБП) подтвердились наличие поликистоза печени и почек, асцит. Наблюдалось увеличение размеров печени с обычной эхогенностью, в паренхиме печени выявлялись множественные тонкостенные кисты с максимальным размером 60 мм. В брюшной полости визуализировалась в значительном количестве свободная жидкость. В почках обнаружены множественные тонкостенные кисты с максимальным диаметром 37 мм.
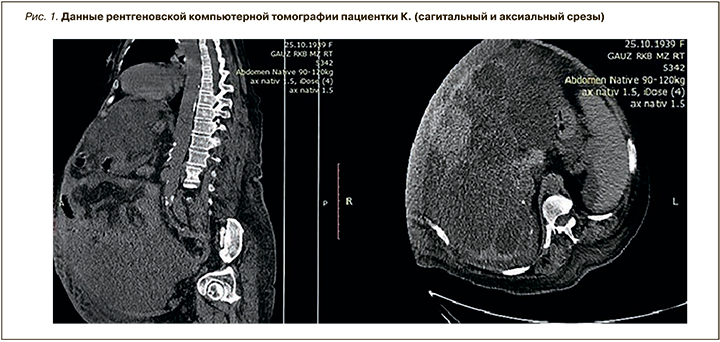
Для визуализации также была проведена рентгеновская компьютерная томография (рис. 1): структура печени неоднородная за счет множественных кист (размер одной из них – 157 мм), с кальцинированными стенками. Контуры почек деформированы, структура неоднородная за счет множественных кист, виден конкремент размером 10 мм. Жидкость в брюшной полости и малом тазу, остальные органы без особенностей.
При эзофагодуоденоскопии (ЭГДС) выявлены варикозно расширенные вены пищевода 1 степени, эрозивный гастрит, дуоденит.
Пациентке был выполнен лапароцентез (забор асцитической жидкости до 20 л с последующим восполнением альбумином).
С учетом всех клинико-лабораторных данных был сформулирован окончательный диагноз «Поликистозная болезнь печени и почек. Синдром портальной гипертензии. Асцит. Лапароцентез (18.06.2021, 23.06.2021). Варикозное расширение вен пищевода 1 степени. Гепатогенная гастропатия: эрозивный гастрит, дуоденит. Мочекаменная болезнь, конкремент чашечки нижней трети левой почки. Острое почечное повреждение (преренальная) на фоне хронической болезни почек (СКФ С4 28 мл/ мин/1,73). Анемия средней степени тяжести смешанного генеза. Гипертоническая болезнь 3 стадии, риск 4. Хроническая сердечная недостаточность 2а, ФК 3».
Пациентка получала следующее стационарное лечение: свежезамороженная плазма – 600 мл внутривенно; альбумин 20% – 50 мл внутривенно № 2; метопролол – 12,5 мг 2 раза/сут; фуросемид – 40 мг/сут; спиронолактон – 50 мг/сут; L-орнитин-L-аспартат – 10 мг/сут; лактулоза – 20 мл/сут; железа гидроксид III валентный – 4,0 мл внутривенно № 5. После проведенного лечения отмечалось незначительное улучшение состояния пациентки.
При выписке пациентке были даны необходимые рекомендации и назначена амбулаторная терапия: урсодезоксихолевая кислота – 250 мг 3 раза/сут; фуросемид – 40 мг/сут; омепразол – 20 мг/сут; препараты железа; амоксициллин + клавулановая кислота – 1200 мг 3 раза/сут; белковые препараты; лактулоза 20 мл/сут. Пациентка была включена в лист ожидания трасплантации печени. Известно, что после выписки через месяц пациентка скончалась.
Из анамнеза удалось выяснить, что у дочери и внучки пациентки также имеется бессимптомный поликистоз печени и почек, что позволило сделать предположение о наследственном характере этого заболевания.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ № 2
Пациентку Н., 53 года, беспокоили жалобы на общую слабость, утомляемость, кожный зуд, тошноту по утрам (без рвоты), тяжесть в правом подреберье после еды, снижение аппетита, уменьшение веса до 5 кг за 3 мес, повышение АД до 150/90 мм рт.ст. Была произведена госпитализация для верификации диагноза и подбора терапии. Диагноз при поступлении «Поликистозная болезнь печени и почек. Желчнокаменная болезнь (Лапароскопическая холецистэктомия в 2017 г.). Гипертоническая болезнь 2 стадия, риск 2. Хроническая сердечная недостаточность 1. ФК 2. Состояние после удаления матки (миома тела матки в 2011 г.). Псориаз. Ожирение I степени».
Данные анамнеза: диагноз поликистоза печени и почек впервые был выставлен пациентке в 2012 г. в ходе лапароскопической холецистэктомии, однако долгое время после этого она не обследовалась. В 2019 г. прошла обследование в Республиканском клиническом онкологическом диспансере (РКОД), на УЗИ ОБП отмечалось увеличение размеров печени, повышение ее эхогенности, с неоднородной структурой за счет множественных кист сливного характера до 115 мм, в левой доле визуализировалась гемангиома. Почки также были увеличены в размерах, с ровными контурами, в паренхиме обеих почек обнаружено множество кист (справа до 40 мм, слева до 68 мм). Осенью 2021 г. пациентка начала отмечать нарастающую слабость, была повторно госпитализирована в РКОД, где ей была выполнена пункция кисты печени под УЗИ контролем; в цитологическом материале обнаружены межклеточное вещество, макрофаги, эритроциты из кистозной полости. У пациентки имелся отягощенный семейный анамнез – поликистоз печени и почек у отца и родной сестры, программный гемодиализ у брата (ХБП 5 по поводу поликистоза почек), что также указывало на наследственный характер заболевания.
Данные объективного осмотра: состояние удовлетворительное, живот безболезненный, печень выступает на 3 см от реберной дуги, бугристая. Размеры по Курлову 13 × 10 × 9 см, симметричная пастозность стоп.
Данные общего анализа крови при дообследовании: повышение СОЭ (39 мм/ч).
Данные биохимического исследования крови: изменения в ФПП (билирубин общий – 29,3 мкмоль/л; билирубин связанный – 22,3 мкмоль/л; АЛТ – 93,4 Ед/л; АСТ – 68,4 Ед/л; ЩФ – 589,8 Ед/л; ГГТП – 373 Ед/л); увеличение С-реактивного белка (5,2 мг/л); показатель церулоплазмина – в пределах референсных значений (40 мг/дл). В протеинограмме гипоальбуминемия (51,3%) с увеличением фракции γ-глобулина (20,9%); альбумин-глобулиновый коэффициент снижен (1,05). Вирусные маркеры гепатита и ВИЧ не выявлены. Показатели коагулограммы в норме. Гормональное исследование щитовидной железы отклонений не выявило.
Общий анализ мочи без особенностей. Дополнительно был исследован ген гистосовместимости HLA-27 (не обнаружен).
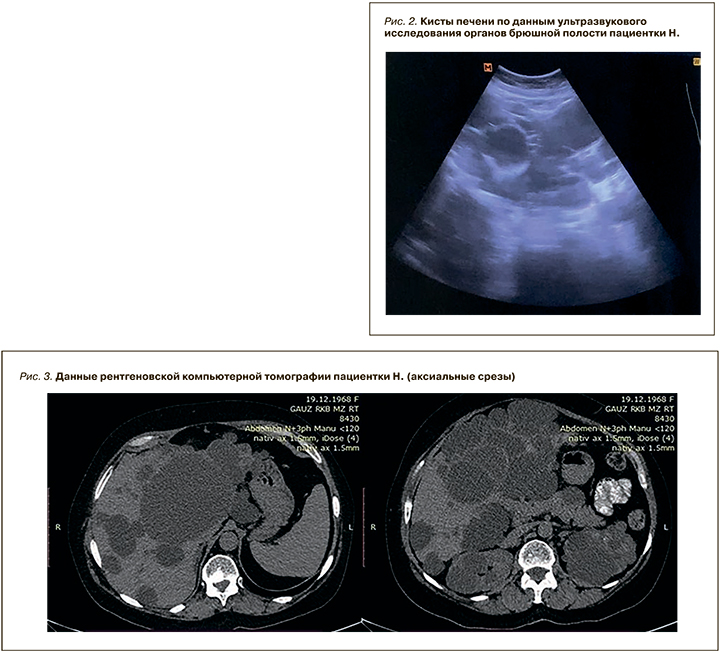
Результаты УЗИ ОБП: размеры печени увеличены за счет множественных тонкостенных образований с максимальным диаметром 95 мм (рис. 2). В брюшной полости свободная жидкость не визуализируется. Обе почки представлены множественными тонкостенными образованиями с максимальным диаметром слева 52 мм, справа – 39мм.
УЗИ щитовидной железы без особенностей.
Ультразвуковая эластография печени выявила признаки поликистоза печени, признаки повышения жидкости паренхимы печени (F2 по шкале Метавир).
Данные рентгеновской компьютерной томографии (рис. 3): структура печени неоднородна за счет множественных жидкостных образований различных размеров, максимально до 122 мм. Обе почки деформированы за счет жидкостных образований с максимальным размером до 56 мм. Свободная жидкость не обнаружена.
Проведена ЭГДС, обнаружены признаки хронического гастродуоденита.
При колоноскопии определены признаки долихосигмы и дивертикулы сигмовидной кишки.
Пациентка была переведена в хирургическое отделение, где ей было произведено повторное УЗИ печени и принято решение о проведении бисегментотомии 3, 4 сегментов печени с последующей постановкой пациентки на очередь для пересадки печени.
С учетом анамнестических и клинико-лабораторных данных при выписке был выставлен диагноз «Поликистозная болезнь печени и почек. Бисегментотомия 3,4 сегментов печени. Хронический поверхностный гастродуоденит, вне обострения. Дивертикулярная болезнь сигмовидной кишки. Лапароскопическая холецистэктомия по поводу желчнокаменной болезни в 2017 г. Гипертоническая болезнь 2 стадии, риск 2. Хроническая сердечная недостаточность 1, ФК 2. Состояние после удаления матки (миома тела матки) в 2011 г. Ожирение 1 степени».
В стационаре пациентка получала лечение: урсодезоксихолевая кислота – 250 мг 3 раза/сут, лозартан + гидрохлортиазид – 50 мг +12,5 мг 1 раз/сут с положительным эффектом.
При выписке пациентке были даны рекомендации, назначена амбулаторная терапия: урсодезоксихолевая кислота – 250 мг 3 раза/сут, лозартан + гидрохлортиазид – 50 мг +12,5 мг 1 раз/сут.
В динамике пациентка отмечала улучшение самочувствия – дискомфорт в правом подреберье и кожный зуд не беспокоил; дети и внуки обследованы – кисты в печени и почках не выявлены.
ОБСУЖДЕНИЕ
Представленные клинические случаи интересны тем, что у описываемых пациентов с ПБП наблюдались клинические и лабораторные проявления болезни. По литературным данным, у большинства больных с ПБП отмечается бессимптомное течение болезни, и лишь малая часть пациентов имеет тяжелые осложнения и нуждается в госпитализации [1]. Со временем у 20% пациентов заболевание проявляется в виде одышки, раннего чувства насыщения, вздутия живота, недоедания, гастроэзофагеального рефлюкса, боли в спине из-за гепатомегалии [4]. Кроме того, у таких больных могут развиться такие серьезные состояния, как портальная гипертензия, асцит, варикозные кровотечения [5]. Действительно, клинический случай № 1 иллюстрирует наличие такого редкого осложнения ПБП, как портальная гипертензия, декомпенсация которой привела к смерти пациентки.
Учитывая клинику, данные лабораторных и инструментальных исследований, отягощенность семейного анамнеза, можно предположить наличие у обсуждаемых пациентов аутосомно-доминантного поликистоза печени с сочетанным поликистозом почек. В первом клиническом примере дебют болезни у пациентки произошел в 76 лет, в примере № 2 – в 44 года. Это согласуется с литературными данными о том, что кисты печени не определяются в раннем возрасте и обычно выявляются в возрасте старше 40 лет. При этом число и размер кист увеличиваются с возрастом [6].
Следует отметить, что оба пациента были женского пола. Из-за аутосомного типа наследования мужчины и женщины должны подвергаться одинаковому риску ПБП, однако, согласно литературным данным, у лиц женского пола заболевание встречается в 6 раз чаще. В научных работах указано, что это связано с более высоким уровнем у женщин эстрогена, который может стимулировать образование кист [7].
Что касается терапии, то большинство бессимптомных пациентов не нуждается в лечении. Для больных с симптомами сдавления выбор лечения зависит от степени выраженности, распространенности и анатомии кист; при этом основным методом выступает хирургическое лечение, которое может включать фенестрацию кисты, резекцию или трансплантацию печени [8]. УЗИ печени и обеих почек в сочетании с обширным семейным анамнезом служит одним из основных этапов при принятии диагностических решений [9].
ЗАКЛЮЧЕНИЕ
ПБП, как правило, является наследственным заболеванием и часто ассоциируется с поликистозом почек. С возрастом возможна прогрессия заболевания, поэтому крайне важен динамический контроль пациентов, также немаловажно проводить скрининг родственников для ранней диагностики. Бессимптомное течение болезни в 20% случаях может прогрессировать, приводя к тяжелым осложнениям, единственным способом лечения которых служит трансплантация печени.
