
Аритмогенная дисплазия правого желудочка (АДПЖ) — первичная кардиомиопатия, первые описания которой представлены G. Fontaine и соавт. в 1977— 1982 гг. [1] как случаи внезапной смерти у молодых людей с правожелудочковой тахикардией без структурных аномалий сердца. Для лечения тахикардии было предложено выполнение вентрикулотомии, при этом определялось истончение мышцы правого желудочка (ПЖ) и ее жировое замещение, что позволило проводить аналогии с болезнью Уля («пергаментное сердце») и обсуждать генетическую природу заболевания [2]. В настоящее время АДПЖ определяется как генетически детерминированное фиброзно-жировое замещение миокарда преимущественно ПЖ, которое проявляется в первую очередь правожелудочковыми нарушениями ритма (экстрасистолией, желудочковой тахикардией — ЖТ) с высоким риском внезапной сердечной смерти.
Установлено, что генетический дефект при АДПЖ заложен на уровне белков десмосом — плакоглобина, десмоплакина, плакофилина, десмоглеина. Хотя основной тип наследования АДПЖ — аутосомно-доминантный, женщины болеют в 3 раза чаще мужчин; распространенность АДПЖ составляет 1 на 2—5 тыс. населения [3, 4]. Заболевание диагностируется главным образом аритмологами и остается практически неизвестным не только широкому кругу врачей, но и специалистам — кардиологам, кардиохирургам, морфологам.
В 1994 г. были сформулированы критерии диагностики АДПЖ [5], которые существенно модифицированы в 2010 г. [6]: наиболее важны количественные признаки дилатации ПЖ с локальными дискинезиями по данным эхокардиографии (ЭхоКГ) и магнитно-резонансной томо графии (МРТ), изменения в правых грудных отведениях на электрокардиограмме — ЭКГ (ε-волна, отрицательные зубцы Т), устойчивая ЖТ из ПЖ, фиброзное либо фиброзно-жировое замещение миокарда и обнаружение специфических мутаций (см. таблицу). При этом пороговое число желудочковых экстрасистол (ЖЭ) снижено с 1000 до 500 в сутки, а обнаружение жира в ПЖ не является обязательным не только при МРТ, но и при морфологическом исследовании.
Наш опыт постановки диагноза АДПЖ включает 15 случаев и позволяет констатировать выраженный клинический полиморфизм этого заболевания. Далее мы представим описание выделенных нами трех основных вариантов АДПЖ с их общей характеристикой и клиническими примерами (диагноз, в большинстве случаев достоверный, к настоящему времени еще не имеет генетического подтверждения и ставился на основании критериев 2010 г.).
Таблица. Европейские критерии диагностики АДПЖ (пересмотр 2010 г.).
Вариант I (типичный) пока преобладает у наших пациентов (в 2 случаях диагноз АДПЖ достоверный, в 6 — вероятный). Подозрение на АДПЖ возникает при этом варианте в связи с обнаружением частой (в среднем 26 тыс./сут) правожелудочковой экстрасистолии (ПЖЭ) в отсутствие структурно-функциональных изменений по данным ЭхоКГ, как правило, у молодых женщин (средний возраст в подгруппе 44,5 года, женщин 75%). Ключом к постановке диагноза становятся данные МРТ,
которая обязательно должна выполняться таким больным: утолщение слоя перикардиального жира по передней стенке ПЖ с участками «наползания» на миокард ПЖ, увеличение конечного диастолического размера (КДР) и расширение выводного отдела ПЖ (ВОПЖ), в среднем 42,7±2,7 и 31,4±3,8 мм, участки истончения и дискинезии передней стенки ПЖ.
Давность аритмического анамнеза составляет в среднем более 10 лет (от 5 до 15), аритмия является ведущим (и, как правило, единственным) симптомом болезни, у ⅔ больных носит прогрессирующий характер и сочетается с пробежками неустойчивой ЖТ. В то же время большие критерии ЭКГ у больных этой категории, как правило, не выявляются, что затрудняет диагностику. Отсутствие пароксизмов устойчивой ЖТ делает аритмию достаточно банальной и заставляет проводить дифференциальный диагноз с широким кругом заболеваний. При этом наиболее сложно дифференцировать латентный (без устойчивой ЖТ) аритмический вариант АДПЖ от миокардита. По данным итальянских авторов, среди больных с поставленным на основании оригинальных критериев
диагнозом АДПЖ при эндомиокардиальной биопсии (ЭМБ) диагноз подтверждается лишь у 50%, а у остальных 50% диагностирован активный миокардит, что определило правильность дальнейшей тактики: среди 14 больных АДПЖ с имплантированными кардиовертерами-дефибрилляторами (ИКД) их срабатывание отмечено у 47%, в то время как у больных миокардитом (без ИКД) ЖТ не развивалась [7].
Уровень антикардиальных антител (ФНЦ трансплантологии и искусственных органов им. акад. В.И. Шумакова) оказался нормальным (1:40—1:80) у 50% больных, что рассматривается нами как характерный признак изолированной АДПЖ. В то же время у других 50% больных с типичным вариантом АДПЖ (n=4) отмечены клинико-анамнестические и лабораторно-инструментальные
признаки миокардита (в том числе наличие вирусного генома и значимое повышение титра антикардиальных антител), который расценивается как вторая болезнь и обусловливает необходимость специфического лечения. У отдельных пациентов длительное наблюдение, оценка эффекта противовоспалительной терапии позволяют дифференцировать АДПЖ и миокардит (либо
выявить их сочетание); в других случаях требуется морфологическая верификация одного или обоих заболеваний, подтверждением чему служат следующие клинические наблюдения.
Больная И., 31 года. Перебои в работе сердца впервые возникли в 25 лет; назначен амиодарон, затем этацизин, на 5-й день приема которого развился эпизод ЖТ с потерей сознания; при электрофизиологическом исследовании (ЭФИ) индуцирована ЖТ из выводного тракта ПЖ. Выполнена радиочастотная аблация, после которой отмечался длительный субфебрилитет (не исключался миокардит). Через 2 года выявлен гипертиреоз, проведено лечение мерказолилом. С 29 лет отмечает нарастание слабости, учащение перебоев в работе сердца, сжимающие боли в области сердца; получала аллапинин, конкор, соталекс, ритмонорм; после отмены терапии зарегистрировано 24 тыс. ПЖЭ/сут. По данным МРТ сердца, КДР ПЖ 40 мм, по передней стенке ПЖ вблизи верхушки определяется участок истончения миокарда с наличием жировых включений с признаками локальной дискинезии около 11 мм; ВОПЖ 37 мм, с гипокинезией передней стенки; уплотнение и утолщение листков перикарда с небольшим количеством жидкости; утолщение слоя эпикардиального жира (послевоспалительные изменения).
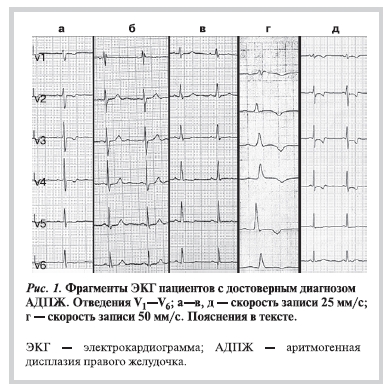
В анализах крови обнаружены антитела к миокарду и стрептококку 1:300 (норма 1:100), к антигенам эндотелия, кардиомиоцитов, проводящей системы — 1:80 (норма 1:40), вирусам герпетической группы. На ЭКГ (рис. 1, а) выявлены неглубокие отрицательные зубцы Т в отведениях V1—V6, которые наряду с клиническими симптомами и признаками поражения перикарда могли расцениваться в рамках миокардита. Выполнена ЭМБ: в субэндокардиальных отделах ПЖ адипозоциты (25—50% площади) и фиброз; миокард разделен фиброзными септами на дольки (выраженное нарушение архитектоники); признаков миокардита нет. Таким образом, верифицирован диагноз АДПЖ; имплантирован ИКД,
возобновлена терапия ритмонормом 600 мг/сут, эффект которого сохраняется в течение 3 лет.
Больная Ж., 69 лет. После 50 лет стали беспокоить перебои в работе сердца, сердцебиение, головные боли, слабость. На ЭКГ изменения не выявлялись. С марта 2011 г. отмечает ухудшение состояния, учащение перебоев, при холтеровском мониторировании (ХМ) ЭКГ зарегистрировано более 20 тыс. ПЖЭ, 27 пробежек неустойчивой ЖТ из 3—4 комплексов QRS. Назначен пропафенон 450 мг/сут, который отменен в связи с брадикардией. На ЭКГ (рис. 1, б) выявлена неполная блокада правой ножки пучка Гиса (ПНПГ), подозрение на ε-волну в отведении V2. При мультиспиральной компьютерной томографии (МСКТ) стенозов коронарных артерий не выявлено. В области модераторного пучка, по передней стенке ВОПЖ, в зоне рядом с предсердно-желудочковой бороздой, в папиллярной мышце левого желудочка (ЛЖ) определялись включения жировой плотности. По данным МРТ, перикард несколько уплотнен, по передней стенке ПЖ — перикардиальные спайки; участки гипо- и дискинезии. ПЖ неравномерно расширен (ВОПЖ 26 мм, КДР 40 мм), преимущественно в приточном отделе; содер-
жание эпикардиального жира повышено до 7—8 мм, с «наползанием» его на миокард ПЖ в области верхушки. МР-сигнал от миокарда ЛЖ однородный.
В анализах крови — ДНК вируса Эпштейна—Барр (полимеразная цепная реакция — ПЦР), IgG к пар-
вовирусу В19, АНФ (с антигеном сердца быка) 1:80 (в норме нет), антитела к эндотелию 1:80, антитела к кардиомиоцитам 1:40, антитела к проводящей системе 1:160. Таким образом, у пожилой больной в отсутствие значимого коронарного атеросклероза выявлено сочетание признаков хронического вирусно-иммунного миокардита и АДПЖ. Проводится противовирусная терапия (ацикловир). Попытка назначения кордарона сопровождалась наряду с подавлением ПЖЭ быстрым урежением синусового ритма до 35 уд/мин, препарат отменен. Больная направлена на имплантацию ИКД (с предварительным ЭФИ).
Вариант II (с правожелудочковой недостаточностью, 5 случаев) характеризуется наличием «необъяснимого» увеличения ПЖ с развитием трикуспидальной регургитации и застоя в большом круге кровообращения и гораздо в меньшей степени отвечает традиционному представлению об АДПЖ: во всех 5 случаях возникли значительные трудности с постановкой диагноза (у 2 больных он верифицирован лишь при аутопсии). Средний возраст составил 36,8 года (22—55 лет), мужчины и женщины болеют одинаково часто (3:2). ПЖЭ (с пробежками ЖТ или без нее) всегда присутствует (среднее количество 4,7 тыс./сут), но может уходить на второй план (особенно в терминальной стадии болезни). Как правило, выявляется также дисфункция ЛЖ без значительного его увеличения (ставятся диагнозы «миокардит», «дилатационная кардиомиопатия» — ДКМП), к которой могут приводить:
1) миокардит (в том числе вирусный); 2) первичное фиброзно-жировое замещение миокарда; 3) сдавление ЛЖ резко увеличенным ПЖ. У всех пациентов имеются большие критерии ЭКГ АДПЖ, что делает диагноз достоверным. Жир в ПЖ или обоих желудочках может выявляться при КТ/МРТ, но может и отсутствовать; типично снижение вольтажа комплексов QRS (в литературе имеются
лишь единичные указания на значение этого признака в диагностике АДПЖ [8]), по-видимому, процессы фиброзирования у больных с декомпенсацией резко преобладают. Приводим клинические наблюдения.
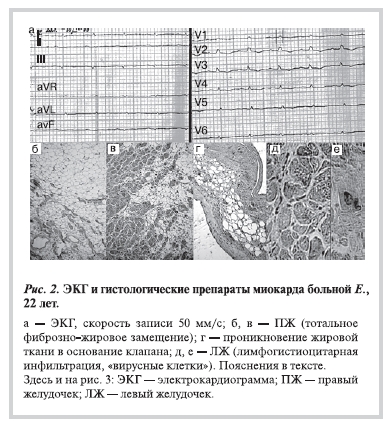
Больная Е., 22 лет. С 20 лет (начало 2009 г.) без видимых причин появилась и нарастала одышка; в РКНПК (по данным ЭхоКГ и МРТ) поставлен диагноз ДКМП. При ХМ ЭКГ регистрировалась ЖЭ. Уже через полгода встал вопрос о пересадке сердца, была направлена в РНЦХ им. акад. Б.В. Петровского, однако от госпитализации отказывалась до конца года, когда состояние резко ухудшилось: значительно наросла одышка, появились отеки и асцит. Была госпитализирована в январе 2010 г. в крайне тяжелом состоянии и через 2 дня умерла при явлениях кардиогенного шока. На ЭКГ (рис. 2, а) — резкое снижение вольтажа комплексов QRS, блокада ПНПГ, отрицательные зубцы Т в отведениях V1—V4. При ЭхоКГ выявлено резкое снижение фракции выброса (ФВ) ЛЖ (17%) при нормальных его размерах (КДР 5,0 см), расширение правых камер сердца с трикуспидальной регургитацией III—IV степени в отсутствие признаков тромбоэмболии легочной артерии и значимой легочной гипертензии.
Наиболее вероятным представлялось развитие тяжелого вирусного миокардита. Действительно, при исследовании миокарда методом ПЦР (ДНК-диагностика) выявлен геном вируса герпеса 6-го типа. Однако при морфологическом исследовании наряду с лимфогистиоцитарной инфильтрацией и единичными «вирусными» клетками в ЛЖ (рис. 2, д, е) выявлено практически тотальное фиброзно-жировое замещение миокарда ПЖ (его площадь местами менее 25%) с образованием соединительно-тканных септ (рис. 2, б, в), прослойки жировой ткани в клапанах (рис. 2, г). Таким образом, причиной
острой декомпенсации сердечной деятельности у больной с АДПЖ в последние месяцы жизни стал, по-видимому, вирусный миокардит. Диагноз АДПЖ был достоверным еще до получения данных аутопсии, однако в отсутствие морфологических данных диагностика АДПЖ, которая сочетается с активным миокардитом, крайне сложна, что подтверждает и следующее наблюдение.
Больной Ч., 40 лет. В возрасте 5 лет оперирован проф. Е.Н. Мешалкиным по поводу врожденного порока сердца (пластика межжелудочковой перегородки (МЖП) синтетической заплатой, инфундибулотомия, инструментальная легочная чрезжелудочковая вальвулодилатация).
С этого времени наблюдались полная блокада ПНПГ, отрицательные зубцы Т в отведениях V1—V4. C 18 лет отмечена наджелудочковая экстрасистолия. В возрасте 23 лет (1993 г.) после острой респираторной вирусной инфекции возникли пароксизмы мерцания-трепетания предсердий, потливость, субфебрилитет; повторный эпизод трепетания купировался через месяц во время ветряной оспы. При ЭхоКГ выявлялось умеренное расширение желудочков. В дальнейшем периодически госпитализировался по поводу нарушений ритма (рис. 3, а). В 2006 г. после острой респираторной вирусной инфекции появились слабость, отеки, субфебрилитет, предобморочные
состояния, выявлены 6 тыс. ЖЭ, короткие пароксизмы ЖТ (частота сердечных сокращений — ЧСС 203 уд/мин), атриовентрикулярная блокада II степени 2-го типа, паузы до 2,4 с, брадикардия до 38 уд/мин, снижение ФВ до 45%. В связи с подозрением на миокардит получал нестероидные противовоспалительные препараты. В 2007 г. в РНЦХ после курса терапии преднизолоном (с субъективным улучшением) имплантирован электрокардиостимулятор (ЭКС) в режиме DDDR (при ЭФИ — инцизионная предсердная тахикардия, ЖЭ четырех морфологий). Назначен амиодарон. Через 1,5 года развился тяжелый тиреотоксикоз со снижением ФВ до 25%, кордарон заменен соталолом, достигнут эутиреоз (преднизолон, тиамазол).
С ноября 2009 г. в связи с частой ЖЭ (7—8 тыс./сут), субфебрилитетом, одышкой, повышением титров антител к эндотелию, проводящей системе (1:160) возобновлена терапия преднизолоном 20 мг/сут, диуретиками. ФВ ЛЖ составляла 34%. При МСКТ определялись повышенная трабекулярность ПЖ, коронарные артерии интактны. Пациент находился на постоянной стимуляции ПЖ в режиме VVI (у больного постоянная мерцательная аритмия), рекомендована замена ЭКС ресинхронизирующим
устройством с функцией дефибриллятора (СRT-D). Для верификации миокардита планировалось проведение ЭМБ, которая по техническим причинам была отложена, в дальнейшем пациент от процедуры отказался. После перенесенного в апреле острого бронхита отмечены нарастание ЖЭ до 11 тыс./сут, появление эпизодов ЖТ продолжительностью до 3 мин (400 за сутки). Была увеличена доза преднизолона, начато насыщение кордароном. Экстрасистолия была частично подавлена, эпизодов устойчивой ЖТ не было. Пациенту рекомендована имплантация CRT-D, в связи с этим планировалось
обследование в Германии. В сентябре 2010 г. поступил в клинику с нарастанием одышки, отеков, возобновлением субфебрилитета (преднизолон к этому времени отменил); через 2 нед внезапно умер.
На ЭКГ (рис. 3, б) определялось практически полное отсутствие зубцов R на фоне постоянной желудочковой стимуляции, при последней ЭхоКГ — КДР ЛЖ 5,3 см, ФВ ЛЖ 24%, митральная регургитация I—II степени, dp/dt 355 мм рт.ст., VTI 8,5 см, левое предсердие 120 мл, правое — 222 мл, ПЖ 3,5 см, ФВ ПЖ 65%, трикуспидальная регургитация III степени, фиброзное кольцо 60 мм, систолическое давление в легочной артерии 41 мм рт.ст. Выраженная трабекулярность ЛЖ (некомпактный миокард?). Несмотря на отсутствие маркеров активной вирусной инфекции за все время наблюдения, диагноз миокардита (с учетом анамнеза, эффективности глюкокортикостероидов) был высоковероятен; перед смертью отмечено повышение антикардиальных антител (специфический АНФ, антитела к проводящей системе 1:160, к эндотелию — 1:320). В то же время врожденный порок
сердца, подозрение на некомпактный миокард, прогрессирующая дилатация правых отделов сердца делали вероятным наличие у пациента генетической кардиомиопатии, в том числе АДПЖ (МРТ не проводилась в связи с наличием ЭКС, при КТ жир не обнаружен).
Рисунок 3. ЭКГ и гистологические препараты миокарда больного Ч., 40 лет.
По данным аутопсии, вес сердца 400 г, гипертрофия стенок (15 мм) и трабекул ЛЖ, кардиосклероз. Миокард ПЖ с очагами фиброза и жировой дистрофии; эндокард плотный, трабекулярность повышена. Методом ПЦР выявлен геном вирусов герпеса 6-го типа (в обоих желудочках) и 1-го типа (в ЛЖ), при морфологическом исследовании: в ЛЖ (рис. 3, в) — выраженные поля фиброза с образованием септ, дистрофия и гипертрофия кардиомиоцитов, интерстициальные и периваскулярные
лимфогистиоцитарные инфильтраты (более 14 клеток при большом увеличении); в ПЖ (рис. 3, г) — участок субэндокардиального фиброза с участками сохраненных кардиомиоцитов, обширные (до 60—70% площади) поля жировой ткани в сочетании с выраженным фиброзом и образованием соединительно-тканных септ; определяются клетки с крупными, деформированными ядрами
и перинуклеарным лизисом цитоплазмы («вирусные»). Таким образом, выявлено сочетание признаков АДПЖ и активного вирусного миокардита.
Критерием АДПЖ является обнаружение не только жира, но и фиброза. По данным итальянских авторов, доля жира в миокарде при ДКМП (0,07%) сопоставима с таковой в контроле (0,33%) и никогда не бывает столь высокой, как при АДПЖ (13,3%): содержание жира в биоптатах более 3,21% с чувствительностью 67% и специфичностью 92% позволяет ставить диагноз АДПЖ [8]. АДПЖ отличается от банальной ДКМП также большей выраженностью фиброза (24,6 и 21,8% соответственно) и меньшим количеством сохранных кардиомиоцитов (47,3 и 63,4% соответственно). Картина, которую мы видели у наших пациентов, не требовала дальнейшей верификации.
Еще у 2 пациентов данной подгруппы мы диагностировали АДПЖ (в одном случае — в сочетании с миокардитом) на основании преобладания недостаточности ПЖ, его дилатации с элементами дискинезии (в отсутствие значимой легочной гипертензии), выявления ПЖ экстрасистолии (1—3,3 тыс./сут), пробежек неустойчивой ПЖТ, ε-волны (см. рис. 1, в), низкого вольтажа комплексов QRS, а также обнаружения жира в обоих желудочках по данным МСКТ (в одном случае подтвержденное, в другом не подтвержденное с помощью МРТ) при неизмененных коронарных артериях. Без достоверных
признаков миокардита сердце пациента отличалось умеренным расширением не только ПЖ, но и ЛЖ (КДР 7,2 см, индекс 2,9 см/м2), в связи с чем ставился диагноз ДКМП. Вместе с тем показано, что при идиопатической ДКМП существует строгая корреляция размеров ЛЖ и степени «отставания» размеров ПЖ: по мере увеличения ЛЖ его объем все в большей степени преобладает над объемом ПЖ [9]. Принципиальным отличием поражения ПЖ при АДПЖ от его вовлечения в патологический процесс при ДКМП является наличие зон дискинезии, локальных аневризм и истончения стенки ПЖ, а также его повышенная трабекулярность. В протокол обследования при подозрении на АДПЖ должно входить определение конечного диастолического объема (КДО), конечного систолического объема ПЖ,
их индексов, ФВ ПЖ и соотношения КДО ЛЖ и ПЖ. В отсутствие дилатации ЛЖ диагноз АДПЖ становится более вероятным.
Наконец, 5-й пациент, 25 лет, перенес в 2003 г. успешную пластику дефекта межпредсердной перегородки; с 2006 г. отметил появление и нарастание перебоев в работе сердца, одышки, отеков; поступил в РНЦХ с тяжелой трикуспидальной недостаточностью, дилатацией правых отделов (по данным МРТ, фиброзная перестройка стенки ПЖ, КДР ПЖ 6,6 см). Данных, подтверждающих
перенесенный инфекционный эндокардит, не выявлено. При ХМ ЭКГ определялись 2,6 тыс. ЖЭ, на ЭКГ
(рис. 1, г) — низкий вольтаж комплексов QRS, расширение их до 0,11 в отведении V1, ε-волна, отрицательные зубцы Т в отведениях V1—V6 (3 больших и 1 малый критерий диагноза АДПЖ).
Вариант III (развернутый) характеризуется наличием рецидивирующих пароксизмов устойчивой
ЖТ и диагностирован нами у 2 пациентов 71 года. Пожилой возраст больных способствовал тому, что
первоначально их состояние расценивалось как ишемическая болезнь сердца (ИБС), большие электрокардиографические критерии АДПЖ оставались нераспознанными.
Больной К., 71 года. ЖЭ отмечается в течение 30 лет, в 1982 г. — первый пароксизм ЖТ с предобморочным состоянием; в связи с наличием на ЭКГ отрицательных зубцов Т состояние расценивалось как ИБС (постинфарктный кардиосклероз). В 1995 г. — повторный эпизод ЖТ, подъем артериального давления до 220/140 мм рт.ст. Подобрана гипотензивная терапия, назначен кордарон. С 1998 г. на фоне нагрузок отмечена рецидивирующая ЖТ; при коронарографии в НЦССХ им. А.Н. Бакулева стенозов не выявлено, имплантирован ИКД; в качестве причины ЖТ рассматривалось гипертоническое сердце (толщина МЖП 14 мм). Выявлены поздние потенциалы желудочков. Отмечались нечастые срабатывания ИКД, в 2004 и 2008 гг. проведена его замена. В 2004 г.
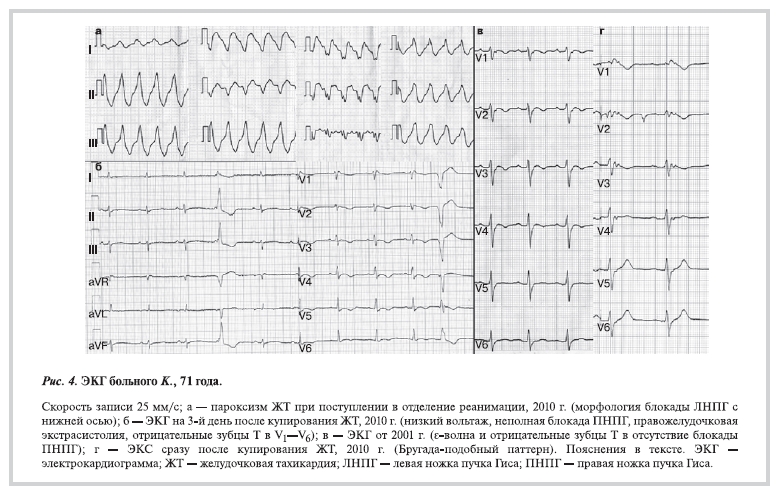
в связи с развитием сильного кашля на фоне бронхиальной астмы кордарон был заменен этацизином (на этом фоне до 3880 ЖЭ/сут). В декабре 2010 г. госпитализирован в отделение реанимации клиники кардиологии с пароксизмом устойчивой ЖТ (рис. 4, а), которая имела морфологию блокады левой ножки пучка Гиса с нижней осью (см. критерии диагноза АДПЖ).
Рисунок 4. ЭКГ больного К., 71 года.
Течение аритмии было необычно тяжелым для пациента с гипертоническим сердцем; кроме того,
ЖЭ и ЖТ появились задолго до развития высокой гипертонии. При ЭхоКГ отмечена диффузная гипертрофия стенок (толщина МЖП 16 мм, толщина задней стенки ЛЖ 14 мм); не исключался диагноз гипертрофической кардиомиопатии. Кроме того, выявлено увеличение ПЖ до 4,8 см. Однако на ЭКГ (рис. 4, б) не было признаков гипертрофии, определялся сниженный вольтаж комплекса QRS, правожелудочковая экстрасистолия и ЖТ, неполная блокада ПНПГ и отрицательные зубцы Т в отведениях V1—V6. При анализе ЭКГ от 2001 г. (рис. 4, в) выявлены (в отсутствие блокады ПНПГ) расширение комплекса QRS до 0,12—0,13 с за счет наличия ε-волны (в отведениях V1—V2, II, III, aVF), отрицательные зубцы Т в отведениях V1—V4. На ЭКГ, зарегистрированной сразу после купирования ЖТ с введением лидокаина и кордарона (рис. 4, г), определялись Бругада-подобные изменения сводчатого типа (характерен для больных с клиническими симптомами) в отведениях V1—V3, что требует
поиска мутаций в гене SCN5A. В литературе встречаются лишь единичные описания сочетания признаков АДПЖ и синдрома Бругада [10]. Таким образом, у пациента выявлены 3 больших и 3 малых критерия АДПЖ.
Наконец, следующее наблюдение. Больная У., 71 года, отмечает частые повторные обмороки, которые расценивались как вазовагальные. При МСКТ коронарные артерии интактны; выявлены жировые включения в миокарде ПЖ. На ЭКГ (рис. 1, д) в отсутствие преходящей блокады ПНПГ определяются ε-волна и отрицательные зубцы Т. Размер ПЖ по данным ЭхоКГ 2,5 см. Планируются проведение МРТ, ЭФИ и решение вопроса об имплантации ИКД. При подтверждении аритмогенного характера обмороков можно констатировать развернутый клинический вариант АДПЖ.
Обсуждение
Еще в 1995 г. G. Fontaine на основании анализа 4 случаев выделил 3 клинические формы АДПЖ [11]: 1) «аритмическая» форма (с нормальной ФВ ЛЖ) — наиболее частый вариант, клинические проявления различны в зависимости от степени дилатации ПЖ; 2) стабильная форма с вовлечением ЛЖ (ФВ от 30 до 50%), при которой сердечная недостаточность (СН) не прогрессирует; 3) прогрессирующая деградация миокарда с развитием бивентрикулярной СН в первые 10 лет после появления симптомов. По данным морфологического исследования умерших, особенно пожилых больных, частота поражения ЛЖ достигает при АДПЖ 100%, однако полное фиброзно-жировое замещение нехарактерно: типичны гипертрофия с участками истончения, атрофия миоцитов, реже встречается некроз [12].
Характерная для многих больных с АДПЖ лимфоцитарная инфильтрация долгое время рассматривалась как вторичный (иммунный?) феномен. Вместе с тем вопрос о взаимоотношениях АДПЖ и миокардита — один из наиболее сложных в этой проблеме. По представлениям ряда авторов, частота трансформации острого миокардита в АДПЖ может достигать 60% [13]. С одной стороны, миокардит способствует реализации аномальной генетической программы; с другой, генетически неполноценный
миокард становится почвой для развития миокардита. Мишенью для выработки аутоантител могут становиться дефектные белки десмосом; нельзя исключить и вторичное происхождение АДПЖ в результате выработки при миокардите антидесмосомных антител (возможно, этим объясняется поздний дебют АДПЖ у ряда наших пожилых больных с миокардитом).
Известно также, что протеаза 2А вирусов Коксаки группы В вызывает повреждение дистрофина [14], а частота обнаружения генома энтеро- и аденовирусов при АДПЖ достигает 75% [15]. Случаи трансформации миокардита в АДПЖ или их сочетание наиболее трудны для диагностики. Описан дебют заболевания в виде миокардита с дисфункцией ЛЖ и последующей трансформацией
в типичную АДПЖ с дилатацией ПЖ и развитием фибрилляции желудочков [16]. В своем обзоре G. Fontaine (2011 г.) уделяет особое внимание миокардиту в клинической картине АДПЖ [17], именно с ним связывая развитие тяжелой СН (в том числе фульминантной), острых эпизодов ЖТ у ряда больных.
По нашим данным, миокардит как самостоятельное заболевание выявляется примерно у 50% больных
с АДПЖ и обусловливает необходимость специфического лечения, в связи с чем мы предлагаем следующую классификацию болезни:
1. Типичная (латентная аритмическая) форма:
А. Изолированная правожелудочковая экстрасистолия
без миокардита.
Б. Неустойчивая ЖТ без миокардита.
В. Изолированная правожелудочковая экстрасистолия/неустойчивая ЖТ с миокардитом.
2. АДПЖ с прогрессирующей СН:
А. Изолированная правожелудочковая недостаточность без миокардита.
Б. Бивентрикулярная недостаточность без миокардита.
В. Бивентрикулярная недостаточность с миокардитом.
3. Развернутая аритмическая форма:
А. Устойчивая ЖТ без дилатации ПЖ без миокардита.
Б. Устойчивая ЖТ с дилатацией ПЖ без миокардита.
В. Устойчивая ЖТ с миокардитом.
По-видимому, заболевание может прогрессировать не только внутри каждой формы, но и от более легкой к более тяжелой форме. И все же можно обсуждать изначальную заданность варианта и тяжести течения, которая определяется, безусловно, генетической природой заболевания у конкретного больного. Например, описаны генетические варианты АДПЖ (мутация гена десмоплакина) с преобладанием поражения ЛЖ [18]; сочетание аритмогенной дисплазии ЛЖ (фиброзно-жировое замещение) с некомпактным миокардом [19]. У нашего пациента с Бругадаподобным паттерном на ЭКГ и относительно ранним появлением агрессивной ЖТ можно предполагать более серьезное, чем у больных с «типичным» вариантом, нарушение процесса реполяризации. Пациенты со II вариантом течения (недостаточность ПЖ) оказались достоверно моложе, чем больные с «типичным» вариантом, у них отмечено более раннее появление больших электрокардиографических критериев, т.е. можно констатировать исходно более тяжелое течение заболевания. Мы надеемся, что проведение ДНК-
диагностики у наших больных с последующим клинико-генетическим анализом позволит объяснить
описанный здесь клинический полиморфизм.
По нашему мнению, предложенная классификация достаточно четко определяет показания к специфическому лечению: только антиаритмические препараты, радиочастотная абляция, ИКД, иммуносупрессивная терапия, трансплантация сердца. Показаниями к ИКД считают (класс рекомендаций IIА, уровень С) перенесенную остановку кровообращения, ЖТ с потерей сознания, полиморфную ЖТ, доказанное выраженное поражение миокарда ПЖ, аневризму ПЖ, вовлечение в процесс ЛЖ (бивентрикулярную форму). Среди наших больных 2 умерли (от пересадки сердца оба
отказывались), 2 имплантирован ИКД, еще у 7 этот вопрос решается; 50% больных получают в качестве антиаритмика амиодарон, всем пациентам с миокардитом (за исключением одной умершей) проводилась или проводится иммуносупрессивная терапия. При этом необходимость назначения амиодарона, появление дисфункции ПЖ и выраженной трикуспидальной недостаточности рассматриваются как факторы неблагоприятного прогноза [20].
Заключение
При диагностике аритмогенной дисплазии правого желудочка необходимо учитывать, что заболевание может встречаться как у мужчин, так и у женщин разного возраста; обнаружение жира при магнитнорезонансной томографии не является обязательным для постановки диагноза, решающее значение имеют оценка состояния правого желудочка и анализ электрокардиограммы. В то же время жир может быть выявлен при мультиспиральной компьютерной томографии (что важно для пациентов с имплантированными устройствами); желудочковые аритмии имеются всегда, но могут уходить на второй план в клинической картине болезни; ведущим клиническим проявлением может быть
«необъяснимая» правожелудочковая недостаточность. Аритмогеная дисплазия правого желудочка сочетается как с другими генетическими кардиомиопатиями, так и с активным вирусным (инфекционно-иммунным) миокардитом (почти у 50% больных); дисфункция левого желудочка связана как собственно с аритмогенной дисплазией правого желудочка, так и с миокардитом и может определять клиническую картину. Для изолированной аритмогенной дисплазии правого желудочка повышение титра антикардиальных антител нехарактерно; биопсия миокарда (с диагностикой вирусной инфекции методом полимеразной цепной реакции) является высокоинформативным методом верификации как аритмогенной дисплазии правого желудочка, так и сопутствующего миокардита.



{kind=link}