
Несмотря на достижения последних десятилетий, диагностика воспалительных заболеваний миокарда по-прежнему вызывает большие трудности, что в первую очередь связано с разнообразием их клинических проявлений. Кроме того, как следует из определения, данного группой экспертов Всемирной организации здравоохранения (1996), миокардит — это, прежде всего, морфологический диагноз, что дополнительно подчеркивает важность проведения эндомиокардиальной биопсии (ЭМБ) для верификации диагноза [1].
Современные представления о патогенезе воспалительных заболеваний миокарда неразрывно связаны с понятием аутоиммунитета. Дополнительным подтверждением аутоиммунной природы миокардита служат такие проявления заболевания, как персистирующая при отсутствии инфекционного агента воспалительная инфильтрация в миокарде, повышение титра циркулирующих кардиоспецифичных аутоантител (ААТ) и в ряде случаев – эффективность иммуносупресивной терапии. При иммуногистохимическом исследовании биоптатов миокарда у больных миокардитом в 59% случаев выявляются специфические ААТ [2]. Вместе с тем циркулирующие антимиокардиальные антитела не являются патогномоничным признаком миокардита и могут определяться у 9—57% больных дилатационной кардиомиопатией (ДКМП), у 9,3% пациентов с острым коронарным синдромом, у 27,6% – с систолической дисфункцией миокарда ишемического генеза и даже у 2—25% практически здоровых лиц [2—4]. Несмотря на низкую специфичность, выявление повышенного титра циркулирующих кардиоспецифических ААТ, как правило, ассоциировано с неблагоприятным прогнозом течения миокардита или ДКМП, а также с более высоким риском развития заболевания у здоровых родственников больных ДКМП [5, 6].
Однако ключевое значение в патогенезе миокардита играет не гуморальный, а клеточный иммунный ответ и, прежде всего, опосредованная главным комплексом гистосовместимости II класса CD4+Т-зависимая лимфоцитарная реакция [7].
В связи с этим особый интерес представляет популяция Т-регуляторных клеток CD4+CD25+, которые участвуют в поддержании иммунологической аутотолерантности и иммунного гомеостаза, подавляя аномальный иммунный ответ.
В какой степени профиль циркулирующих кардиоспецифических ААТ и количество Т-регуляторных клеток CD4+CD25+ отражают тяжесть морфологических и иммуногистохимических изменений у больных лимфоцитарным миокардитом и как это связано с клиническими проявлениями заболевания? На эти вопросы мы попытались ответить в настоящей работе.
Цель исследования: оценить клиническое и прогностическое значение циркулирующих кардиоспецифических ААТ и Т-регуляторных клеток CD4+ у больных миокардитом.
Материал и методы
Исследование проводилось на базе ФГБУ «СЗФМИЦ им. В.А. Алмазова» с сентября 2011 по ноябрь 2013 г. и носило проспективный характер. Обследованы 47 пациентов с лимфоцитарным миокардитом, документированным на основании анализа гистологических и иммуногистохимических данных в соответствии с утвержденными положениями Рабочей группы по заболеваниям миокарда и перикарда Европейского общества кардиологов [8]. В исследуемую группу вошли 18 больных активным и 29 пациентов с пограничным лимфоцитарным миокардитом.
Контрольную группу составили 30 практически здоровых лиц, доноров крови (17 мужчин, 13 женщин, средний возраст 33,8±9,3 года).
Исследование было одобрено этическим комитетом ФГБУ «ФМИЦ им. В.А. Алмазова». Перед включением в исследование все пациенты подписывали информированное согласие. Клиническая характеристика групп представлена в табл. 1.
Всем пациентам проводили ЭхоКГ на аппарате Vivid 7 (GE, США), определяли размеры камер сердца, толщину межжелудочковой перегородки в диастолу, толщину задней стенки левого желудочка (ЛЖ) в диастолу, фракцию выброса (ФВ) ЛЖ по Симпсону.
Биоптаты миокарда фиксировали 10% забуференным формалином. Парафиновые срезы 2—3 мкм окрашивали гематоксилином и эозином по ван Гизону и азур-эозином для оценки воспалительных инфильтратов. Дополнительно выполняли ШИК-реакцию для исключения гликогенозов, а также окраску конго красным и окраску по Перлсу для исключения амилоидоза и гемохроматоза у пациентов старше 50 лет [9].

Для выявления воспалительных изменений в миокарде проводили иммуногистохимическое исследование с антителами к HLA-DR (clone LN3, Leica, 1:300), CD-3 (поликлональные антитела, DAKO, 1:800), CD45 (clone 2B11+PD7/26, DAKO, 1:1200), CD-68 (clone PG-M1, DAKO, 1:1000). Экспрессию HLA-DR оценивали в баллах: 1 балл — экспрессия антигена на единичных клетках воспалительного инфильтрата; 2 балла — на всех клетках инфильтрата; 3 балла — на всех клетках инфильтрата и на эндотелии некоторых сосудов; 4 балла — на всех клетках инфильтрата, на эндотелии всех сосудов и вдоль всех капилляров. Активный миокардит диагностировали при наличии некроза кардиомиоцитов и воспалительного инфильтрата, включающего не менее 14 лейкоцитов на 1 мм2, из них не более 4 моноцитов и ≥7 Т-лимфоцитов CD3+ на 1 мм2 [8].
С помощью стандартизованных иммуноферментных тест-систем группы ЭЛИ-ТЕСТ в сыворотке крови пациентов анализировали профили иммунореактивности маркерных кардиотропных ААТ, направленных к антигену цитоскелета кардиомиоцитов CoM-02, цитоплазматическому антигену кардиомиоцитов CoS-05-40, антигену NO-синтетазы (NOS) и коллагену. Все белковые антигены выделяли на базе лаборатории МИЦ «Иммункулус» с помощью общепринятых хроматографических методов. Кроме того, определяли содержание ААТ к двум пептидным фрагментам белка — переносчика адениновых нуклеотидов (ANT-1 и ANT-2) – белку р53, миозинсвязывающему белку MyBP, М2-холинорецепторам и фрагменту внеклеточной петли β1-адренорецептора. Синтез пептидных фрагментов белков был выполнен компанией «Peptide 2.0 Inc» (Chantilly, США). Пептидный фрагмент внеклеточной петли β1-адренорецептора предоставлен проф. С.Н. Покровским (Институт клинической кардиологии им. А.Л. Мясникова, Москва). Среднюю индивидуальную иммунореактивность сывороток и нормализованное содержание ААТ к каждому из антигенов оценивали согласно инструкции производителя, используя компьютерную программу, прилагаемую к тест-наборам. В работе оценивали индивидуальные особенности «профилей» (паттерн интегральной аутореактивности пациента) [10—12].
Число циркулирующих Т-лимфоцитов CD4+CD25+ и CD4+CD25+FoxP3+ оценивали методом проточной цитометрии с использованием моноклональных антител к CD4, меченных флюоресцеин-изотиоциантом (FITC, R&D™), CD25 фикоэритрином (PE, R&D™) и FoxP3 аллофикоцианином (АРС, R&D™).
Уровень высокочувствительного С-реактивного белка определяли иммунотурбидиметрическим методом, тропонина I — иммуноферментным методом. Для оценки концентрации N-концевого мозгового пропептида (NT-proBNP) в сыворотке крови применяли электрохемилюминесцентный метод с использованием стандартных тест-систем и анализатора.
Статистическую обработку данных проводили с использованием пакета прикладных программ Statistica версия 10.0. Статистический анализ полученных результатов проводили с помощью методов непараметрической статистики (критерии Манна—Уитни, Крускала—Уоллиса. χ2-критерий). Результаты представлены в виде среднего арифметического значения (М), стандартного отклонения (σ) или медианы (Ме) и первого и третьего квартилей (Q25; Q75), а также числа признаков в группе (n). Для анализа комбинированной конечной точки (сердечно-сосудистая летальность и потребность в трансплантации сердца) за начало отсчета была принята дата выполнения ЭМБ, подтверждающей диагноз миокардита, а за окончание — дата смерти пациента или дата трансплантации сердца. Выживаемость пациентов с миокардитом анализировали с помощью кривых Каплана—Мейера. Различия считали статистически значимыми при p<0,05.
Результаты
В дебюте заболевания у 23% пациентов с морфологически документированным миокардитом встречался болевой синдром (кардиалгии или ангинозоподобные боли). С симптомами прогрессирующей сердечной недостаточности (СН) к врачу обратились 60% обследованных больных (см. табл. 1). Желудочковые и наджелудочковые нарушения ритма регистрировались у 86% больных, в том числе в 55% случаев – опасные для жизни желудочковые нарушения ритма: желудочковая тахикардия или фибрилляция желудочков.
У 60% пациентов с миокардитом выявлено повышение уровня тропонина I в сыворотке крови — 1,08 (0; 0,299) нг/мл. Уровень С-реактивного белка у обследованных пациентов варьировался от 1,83 до 10,8 мг/л.
Диагноз миокардита по результатам ЭМБ верифицирован на основании Далласских критериев и критериев Ассоциации сердечно-сосудистых патологов Европы [8, 13]. Согласно Далласским критериям, у 18 пациентов имелся активный миокардит и у 29 — пограничный миокардит (табл. 2). При гистологическом исследовании у 32 больных выявлены фиброзные изменения в миокарде, что свидетельствовало о хронизации воспалительного процесса.
На выраженность воспалительных изменений указывало число клеток воспаления (CD3+, CD45+, CD68+) на единицу площади миокарда (мм2). Достоверных различий между активным и пограничным миокардитом по числу клеток воспаления, инфильтрирующих миокард, не получено. Высокая экспрессия антигена HLA-DR отмечена как в группе больных активным, так и пограничным миокардитом.
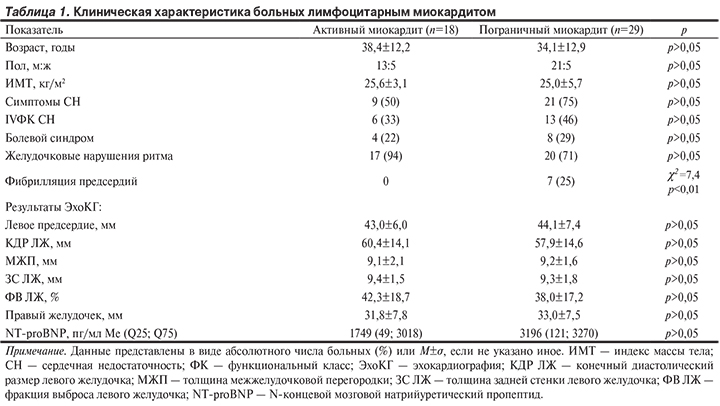
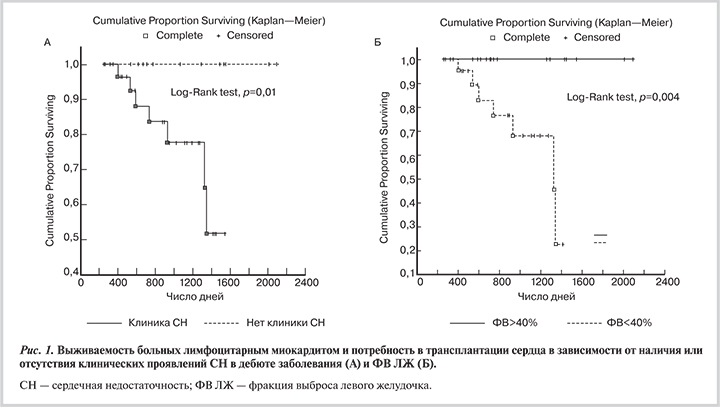
Среди факторов, определяющих прогноз больных лимфоцитарным миокардитом, ключевое значение имели наличие клинически выраженной СН в дебюте заболевания (рис. 1), а также степень снижения ФВ ЛЖ. У больных с ФВ ЛЖ более 40% частота фатального исхода и потребностьв трансплантации сердца были достоверно ниже (p=0,004). При анализе комбинированной конечной точки (сердечно-сосудистая летальность и потребность в трансплантации сердца) выявлена тенденция, свидетельствующая о том, что прогноз у пациентов с пограничным миокардитом был хуже, чем у пациентов с активным воспалительным процессом (рис. 2). Подтверждением этому служили и более высокие концентрации мозгового натрийуретического пропептида в сыворотке крови у пациентов с пограничным миокардитом.
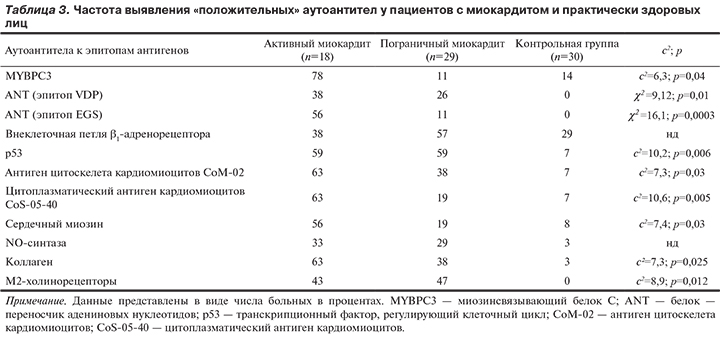
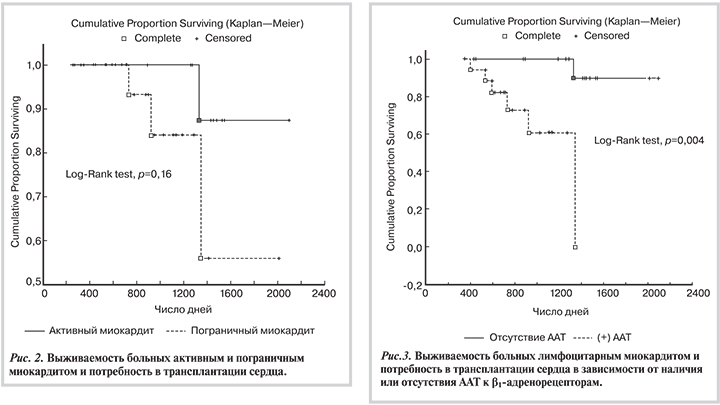
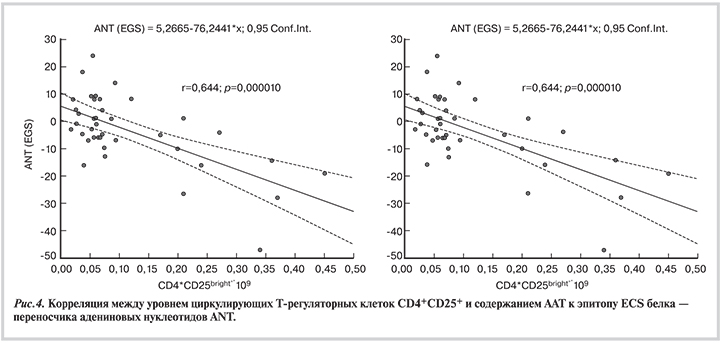
Уровень ААТ в сыворотке крови может быть ранним и весьма специфическим маркером возникновения патологического процесса в том или ином органе. Прямым доказательством этого могут быть результаты настоящего исследования, в соответствии с которыми пациенты с миокардитом имели специфический по сравнению с группой практически здоровых доноров крови «профиль» ААТ (табл. 3). Отличительной особенностью больных активным миокардитом было появление пиков иммунореактивности ААТ к саркомерным, цитоскелетным и цитоплазматическим белкам кардиомиоцитов, а также пиков ААТ к различным эпитопам белка — переносчика адениновых нуклеотидов ANT, свидетельствующих о наличии митохондриальных нарушений в клетках миокарда. Напротив, у больных пограничным миокардитом чаще повышался уровень ААТ к β1-адренорецепторам, что, вероятно, связано с большей частотой как клинически выраженной СН, так и нарушений ритма сердца. Более того, повышенный уровень ААТ к β1-адренорецепторам был независимым предиктором неблагоприятного исхода у больных лимфоцитарным миокардитом (рис. 3). На наличие повреждения миокарда как в группе больных активным (44%), так и в группе пациентов с пограничным миокардитом (26%) указывало появление пиков иммунореактивности ААТ к проапоптотическому белку р53.
Однонаправлено в обеих подгруппах менялся и уровень ААТ к NO-синтазе, свидетельствующий о развитии дисфункции эндотелия у больных миокардитом. Выявлено, что повышение уровня циркулирующих кардиоспецифических ААТ может отражать патологическое ремоделирование миокарда у пациентов с воспалительными заболеваниями. В частности, повышенный уровень ААТ к сердечному миозину был ассоциирован с увеличением конечного систолического размера ЛЖ (р=0,02), а повышение уровня ААТ к коллагену — со снижением ФВ ЛЖ (rs=-0,446; p=0,008). Появление кардиоспецифических ААТ может быть не только отражением патологического процесса.
Нельзя исключить, что ААТ в ряде случаев выполняют защитную функцию. Например, между величиной ФВ ЛЖ и содержанием ААТ к цитоплазматическому антигену кардиомиоцитов CoS-05-40 (rs=0,419; p=0,014) и NO-синтазе (rs=0,340; p<0,05) выявлена положительная корреляция.
В многочисленных клинических и экспериментальных исследованиях показано, что между развитием воспалительных изменений в миокарде и активацией Т-регуляторных клеток существует обратная связь. В отличие от традиционных представлений о том, что число Т-регуляторных клеток снижается при развитии аутоиммунных заболеваний, в настоящем исследовании у больных лимфоцитарным миокардитом выявлено увеличение популяции клеток CD4+CD25+ и клеток CD4+CD25bright+FOXP3+ по сравнению с контрольной группой.
Рост популяции Т-регуляторных клеток CD4+CD25+ ассоциирован с повышением концентрации NT-proBNP, который служит маркером тяжести и прогноза у больных СН (rs=0,426; p=0,019). Наряду с этим выявлена обратная корреляция между содержанием ААТ к эпитопу ECS белка – переносчика адениновых нуклеотидов ANT, кардиоспецифичному миозину и уровнем циркулирующих Т-регуляторных клеток CD4+CD25+ (β=-0,644; p=0,00001 и rs=-0,361; p=0,026 соответственно) (рис. 4).
Обсуждение
В патогенезе воспалительных заболеваний миокарда наряду с прямым повреждающим действием инфекции или токсического фактора и продукцией провоспалительных цитокинов, важную роль отводят нарушениям клеточного и гуморального аутоиммунитета. Доказательством участия CD4+ Т-лимфоцитов в патогенезе воспалительных заболеваний миокарда служит ставшая классической модель экспериментального аутоиммунного миокардита, в которой показано, что снижение уровня Т-клеток CD4+ препятствует развитию миокардита, а введение этих клеток от заболевшей особи здоровым животным способствует его возникновению [14]. Таким образом, CD4+ Т-клетки — необходимое и достаточное условие для развития миокардита [7]. Функциональное и фенотипическое разделение CD4+ T-клеток осуществляют по характеру секретируемых цитокинов на так называемые клетки Th1 и Th2 в зависимости от продукции ими интерферона γ (ИФН-γ) и интерлейкина-4 (ИЛ-4) соответственно [15].
Существуют убедительные доказательства как в эксперименте, так и в клинике участия ИФН-γ и клеток Th1 в патогенезе миокардита, вызванного кардиотропными вирусами [16, 17]. Вместе с тем блокада продукции ИФН-γ приводила не к подавлению воспалительных изменений в миокарде, а к их парадоксальной активации, вплоть до развития тяжелой СН [18]. Дело в том, что в ряде исследований продемонстрированы защитные функции ИФН-γ, в том числе в отношении контроля апоптоза ауторегуляторных Т-лимфоцитов [19].
Впервые Т-регуляторные клетки охарактеризованы S. Sakaguchi и соавт. в середине 90-х гг. XX века как популяция клеток CD4+, дополнительно экспрессирующих рецептор ИЛ-2 CD25 [20]. Позднее в Х-хромосоме был найден ген FOXP3, экспрессия которого связана с продукцией фактора, необходимого для контроля развития и функционирования Т-регуляторных клеток [21, 22]. Несмотря на то что механизмы, вызывающие экспрессию FOXP3, хорошо изучены, четких представлений о том, каким образом контролируются Т-регуляторные клетки, пока нет. В последние годы получены доказательства, что иммунорегуляторная функция этих клеток осуществляется благодаря продукции ингибиторных цитокинов, таких как ИЛ-10 и трансформирующий β-фактор роста, конкуренции за ИЛ-2, а также за счет контактного взаимодействия клеток [23, 24].
Данные о представительстве Т-регуляторных клеток в периферической крови больных аутоиммунными заболеваниями широко варьируются и во многом зависят от фенотипических маркеров, которые используются для характеристики этих клеток. На сегодняшний день FOXP3 рассматривается как наиболее специфический внутриклеточный маркер Т-регуляторных клеток. В клинических исследованиях, включавших больных сахарным диабетом 1-го типа, системной красной волчанкой, ревматоидным артритом и рассеянным склерозом, продемонстрировано как снижение числа циркулирующих Т-регуляторных клеток СD4+CD25+, так и отсутствие принципиальных различий по сравнению с контролем [25]. Не выявлено корреляций между темпами прогрессирования периферического и коронарного атеросклероза и числом циркулирующих CD4+CD25+ и экспрессией мРНК FOXP3 [26]. Напротив, в нашем исследовании выявлено увеличение числа циркулирующих Т-регуляторных клеток CD4+CD25+ и CD4+CD25bright+FOXP3+, что, вероятно, носит компенсаторный характер. Данный факт подтверждается наличием положительной корреляции между представительством клеток CD4+CD25+ и уровнем NT-proBNP. Однако повышение уровня циркулирующих ААТ было ассоциировано с первичным или вторичным снижением числа и/или нарушением функции Т-регуляторных клеток.
Повреждение миокарда служит пусковым механизмом развития аутоиммунных реакций при сердечно-сосудистых заболеваниях, ключевым звеном которых являются феномен «молекулярной мимикрии» и перекрестная иммунореактивность. Кардиоспецифические ААТ могут оказывать дополнительное воздействие на структурно-функциональные изменения в сердце. В частности, ААТ к β1-адренорецепторам могут индуцировать апоптоз кардиомиоцитов, тем самым способствуя дилатации полостей сердца [27]. Хотя наличие ААТ к β1-адренорецепторам ассоциировано с высоким риском развития и прогрессирования СН, четкие данные о распространенности, частоте появления и кинетике этих ААТ в периферической крови пациентов с СН различного генеза по-прежнему отсутствуют. Решить существующую проблему предполагается в рамках многоцентрового клинического исследования ETiCS, начавшегося в 2010 г. [28].
В настоящем исследовании повышенный уровень ААТ к β1-адренорецепторам зарегистрирован у 58% больных пограничным миокардитом, что согласуется с ранее опубликованными данными отдаленного прогноза у больных активным и пограничным миокардитом, в которых пограничный миокардит ассоциировался с большей дилатацией ЛЖ [29].
Как правило, появление ААТ к β1-адренорецепторам сопряжено с ростом уровня аутоантител к М2-холинорецепторам. Последние оказывают негативные хроно- и инотропное действия, а также ингибирующее влияние на активность аденилатциклазы [30]. Появление аутоантител к М2-холинорецепторам ассоциировано со снижением глобальной сократительной способности ЛЖ у больных ДКМП, что способствует прогрессированию СН [31].
Роли антимитохондриальных ААТ при различных заболеваниях посвящены многочисленные исследования. Появление ААТ к белку — переносчику адениновых нуклеотидов ANT, у больных ДКМП впервые продемонстрировано в работе Н.Р. Schultheiss и соавт. [32]. Дело в том, что аминокислотная последовательность ANT до некоторой степени повторяет последовательность белка энтеровируса Коксаки В3 — одного из основных кардиотропных вирусов, приводящих к развитию миокардита [33]. На экспериментальной модели аутоиммунного миокардита показано, что при нейтрализации циркулирующего ИЛ-17 уровень ААТ к ANT снижается. Высказано предположение, что возможный протективный эффект, связанный с подавлением воспаления, ассоциированного с ИЛ-17, реализуется за счет ингибирования пролиферации В-клеток CD19+ и уменьшения продукции ААТ к ANT [34, 35]. Подтверждением данной гипотезы служат данные о снижении уровня ААТ к ANT в зависимости от тяжести поражения миокарда, полученные в настоящем исследовании.
Аутоиммунитет можно рассматривать как важнейший инструмент удаления поврежденных собственных клеток и вредных продуктов обмена, что обеспечивает перманентную продукцию естественных ААТ у каждого здорового человека [36]. Поэтому синтез ААТ определенной специфичности компенсаторно повышается при возникновении первичного патологического процесса в определенном органе или ткани. Такие саногенные аутоиммунные реакции обеспечивают локальную активацию клиренса пораженного органа. Поэтому повышение уровня циркулирующих ААТ может носить защитную функцию.
Эту гипотезу подтверждают результаты настоящего исследования о более высоких значениях ФВ ЛЖ у больных миокардитом и повышенным уровнем циркулирующих ААТ к цитоплазматическому антигену кардиомиоцитов, а также данные A.O. Doesch и соавт. об улучшении выживаемости больных ДКМП, имеющих повышенный уровень ААТ к тропонину I [37].
Таким образом, результаты настоящего исследования подтверждают, что нарушения в системе аутоиммунитета играют ключевую роль не только в патогенезе, но и в прогнозе воспалительных заболеваний миокарда. Вместе с тем изменения профиля циркулирующих кардиоспецифических ААТ и Т-регуляторных клеток могут носить защитную функцию, что нуждается в дальнейшем изучении.
Авторы выражают искреннюю благодарность профессору А.В. Полетаеву за помощь в организации исследования профиля аутоантител.
Работа выполнена в рамках государственного задания ФГБУ «СЗФМИЦ им. В.А. Алмазова» «Метаболомные и транскриптомные маркеры развития фиброза миокарда».


