
Важнейшей детерминантой реакции крови на повреждение стенки сосуда является активация системы гемостаза, которая обеспечивает не только формирование тромба и закрытие дефекта стенки, но и включение воспалительной реакции, а в ряде случаев — индукцию системного ответа на повреждение [1, 2]. Многоуровневая организация и многофакторность регуляции системы гемостаза определяют широкую вариабельность проявлений патологии системы свертывания крови и патогенетических механизмов ее развития. Изучению гемостаза посвящено огромное количество работ, позволивших описать клинические проявления, основы молекулярной биологии и генетики геморрагических заболеваний, а также определить тактику и стратегию коррекции нарушений гемостаза [3—8]. В то же время анализ патогенеза различных заболеваний внутренних органов (коронарная патология, острое нарушение мозгового кровообращения, гломерулопатии, язвенная болезнь и ее осложнения — острые кровотечения и др.) привел к пониманию участия и роли системы гемостаза в регуляции репаративной регенерации органов [9—11]. В частности, в развитии дизрегенераторного синдрома, ассоциированного с нарушением течения воспаления, ангиогенеза и репарации тканей [12—14]. Так, доказана роль факторов системы гемостаза (пурины, тромбин, СD40 и пр.) в развитии атеросклероза и атеротромбоза, сопровождающегося поражением коронарных и церебральных сосудов; подтверждено участие тромбина, тромбоксанов, Р-селектина и других рецепторов тромбоцитов в патогенезе гломерулопатий; обсуждаются механизмы нарушения межмолекулярных связей в системе коагуляции и функционировании тромбоцитов при кровотечениях разной локализации [15—18].
В настоящее время современная стратегия диагностики нарушений в системе свертывания крови базируется на комплексном анализе всех звеньев регуляции гемостаза. Такой анализ включает, как правило, оценку реологических свойств крови, показателей реактивности сосудистой стенки (эндотелий- и адренозависимая регуляция), адгезии и агрегации тромбоцитов [19—24]. При длительном патологическом процессе, сопровождающемся нарушениями гомеостаза, важная роль отводится параметрам функционального состояния тромбоцитов: спонтанная и индуцированная агрегация, уровень фактора Виллебранда (ФВ), метаболический гомеостаз тромбоцитов. При анализе системы плазменной коагуляции учитываются механизмы реализации внешнего и внутреннего путей. Причем изучение факторов коагуляции имеет смысл лишь при одновременной оценке системы антикоагуляции плазмы крови, учитывающей состояние физиологических антикоагулянтов (антитромбина ІІІ, белка С, тромбомодулина, ингибитора внешнего пути активации свертывания крови). И наконец, судить об эффективности гемостаза нельзя без анализа состояния системы фибринолиза (ключевыми показателями которого являются характеристика фибринолиза, уровень ингибитора активатора плазминогена и пр.) [21—23].
Разнонаправленность такой диагностики породила проблемы анализа и корректной трактовки статуса гемостаза. Подтверждением этого могут быть современные, порой противоречивые, представления о состоянии системы гемостаза при кровотечениях из язв гастродуоденальной зоны. Так, у больных с данной патологией отмечается как усиление (и склонность к тромбообразованию), так и снижение свертывания крови (соответственно, регистрируется уменьшение или увеличение времени рекальцификации плазмы, изменение параметров протромбинового времени). Кроме того, выявлено нарушение толерантности плазмы крови к гепарину (отражающей изменения уровня антитромбина 3), активация фибринолитической системы за счет мощного освобождения активаторов фибринолиза (тканевых киназ, плазминогена и его активатора — урокиназы), изменение функционального статуса тромбоцитов [7, 13, 17].
С нашей точки зрения, для решения данной проблемы требуется не только адекватная оценка статуса системы тромбогенеза, но и анализ возможных механизмов нарушения иммунологической и нейрогуморальной реактивности, компенсаторных реакций сосудистой стенки. Столь широкий спектр требований не случаен, ведь в конечном итоге эффективность гемостаза зависит от сопряженной активации тканевых факторов и стенки сосудов, коагуляционной системы крови и функции тромбоцитов.
В этом отношении оптимальной моделью для анализа системы тромбогенеза являются тромбоциты, что обосновывается рядом положений. Во-первых, тромбоциты представляют собой интегральную часть физиологического ответа на повреждение сосудистой стенки. Во-вторых, тромбоциты могут модулировать статус разных звеньев системы коагуляции, эндотелия и соединительной ткани (система факторов роста). В-третьих, тромбоциты являются триггерами воспалительного ответа в месте повреждения (за счет экспрессии Р-селектина, СD40, освобождения серотонина и гистамина, тромбоксана А2 — ТХА2 и пр.). И в-четвертых, являются мишенью системных и локальных регуляторов, а значит, могут отражать реактивность организма [2, 12, 17].
Механизм активации тромбоцитов
Возможность использования тромбоцитов как модели для анализа реактивности и сосудистого гомеостаза также определяется универсальностью механизмов активации, реализация которых сопровождается повышением концентрации цитозольного Са2+. Ответ тромбоцитов включает активацию интегринов и освобождение вторичной волны медиаторов — аденозиндифосфата (АДФ) и ТХА2, усиливающих прокоагулянтную активность. Источником ионов Са2+ для стимуляции тромбоцитов является как внутри-, так и внеклеточный Са2+. Внутриклеточный Са2+ освобождается из эндоплазматической сети (ЭС) при участии инозитол-3-фосфата (И3Ф), образующегося при участии фосфолипазы С (ФЛС) [7]. Ключевым механизмом входа внеклеточного Са2+ является зависимый от депо Са2+ вход (store-operated Ca2+ entry — SOCE), контролируемый запасом ионов Са2+ в ЭС [25]. В частности, снижение содержания Са2+ в ЭС является триггером для активации кальциевых каналов. Этот механизм в литературе описан как «изнутри наружу» (inside—out) и предполагает первичное повышение уровня внутриклеточного Са2+ за счет выхода из ЭС с последующим открытием кальциевых каналов плазмолеммы и входом извне ионов Са2+.
Освобождение Са2+ из внутриклеточных депо регулируется различными агонистами, и, как правило, сопряжено с активацией изоформ ФЛС, которые гидролизуют фосфоинозитиддифосфат до И3Ф и диацилглицерола. Таким образом, ключевым участником реакций, ведущих к повышению внутриклеточного Са2+, являются ФЛС и запускаемый при этом каскад мессенджеров и трансдукторов. В тромбоцитах описано 3 изоформы фермента: ФЛСb, ФЛСγ и ФЛСd. Доминирующими считаются ФЛСb2/3 и ФЛСγ. Первая регулируется со стороны рецепторов, ассоциированных с белком Gq, тогда как ФЛСγ запускается при активации рецепторов к коллагену — гликопротеин (GP)VI, интегриновыми рецепторами — α2b1 и αІІbβ3, недавно идентифицированным лектиноподобным рецептором тромбоцитов, рецептором ФВ — комплексом GPIb—V—IX и тромбоцитарным рецептором Fcγ IIA (Fcγ IIA). Кроме того, стимуляция ФЛСγ возможна со стороны фосфатидилинозитол-3-киназы (Ф3К), активируемой в нисходящем сигнальном потоке рецепторов, связанных с белком Gi [26, 27].
Активация ФЛС и образование И3Ф ведут к освобождению Са2+ за счет формирования комплекса И3Ф-рецептор. Последний представлен тремя изоформами, среди которых в тромбоцитах доминирующими являются И3Ф—R1 и И3Ф—R2. Регуляция этих рецепторов возможна путем фосфорилирования различных протеинкиназ. Ингибиторный эффект фосфорилирования рецепторов к И3Ф описан при повышении уровня цАМФ и цГМФ. Основным депо Са2+ в тромбоците являются цистерны ЭС, в мембраны которой вмонтированы две изоформы саркоплазматической/эндоплазматической Са2+-АТФазы (SERCA). Причем изоформа SERCA2b имеет массу 100 кД и чувствительна к трипсигаргину, и соответствует системе плотных трубочек — аналогу ЭС тромбоцитов. Вторая изоформа — SERCA3 имеет молекулярную массу 97 кД, экспрессируется в «кислых» органеллах, имеет низкое сродство к трипсигаргину, но чувствительна к 2,5-ди- (t-бутирил)1,4-гидроксиквинону (TBHQ) [25].
Сопряженность выхода ионов Са2+ из депо с открытием кальциевых каналов плазмолеммы связана с работой ряда молекул. Одной из них является трансмембранный белок STIM1 (stromal interaction molecule 1), который одним концом встроен в мембрану ЭС и чувствителен к уровню Са2+. Снижение уровня Са2+ в цистернах ЭС тромбоцита ведет к активации и конформационным изменениям STIM1, которая, стыкуясь с Orai1 (CRACM — calcium release activated calcium modulator) в плазмолемме, способствует открытию ее кальциевых каналов. При дефекте данной молекулы нарушается повышение уровня Са2+, что ограничивает активацию ФЛСb и ФЛСγ, хотя сохраняется стимуляция αІІbβ3 интегрина [21, 27].
Следовательно, стимуляция функционального ответа тромбоцитов зависит от внутриклеточных механизмов, контролирующих уровень Са2+, но определяется связью рецепторов тромбоцитов с различными лигандами. В этом контексте нужно отметить, что тромбоциты являются уникальной биологической системой, несущей огромное количество рецепторов к системным и тканевым регуляторам на динамично ремоделируемой плазмолемме.
Молекулярные основы реализации функционального ответа тромбоцитов
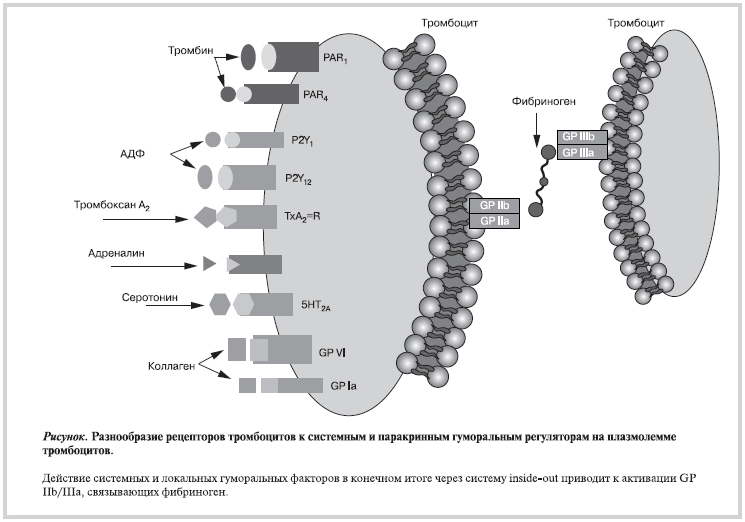
На поверхности тромбоцитов представлен широкий спектр рецепторов, участвующих не только в контроле гемостаза, но и в реализации других функций тромбоцитов, включая регуляцию воспаления, репарации, опухолевого роста и метастазирования (рис.). Рецепторы тромбоцитов многообразны не только по лигандам, но и по химическому составу, и включают следующие [18, 29]:
• многочисленные интегрины (αIIbβ3, α2b1, α5β1, α6β1, αVβ3);
• рецепторы, богатые лейцином (LRR);
• гликопротеины (GPIb—V—IX, Toll-подобные рецепторы);
• рецепторы, ассоциированные с белком G (рецепторы PAR-1 и PAR-4 к тромбину, рецепторы P2Y1 и P2Y12 к АДФ, рецепторы TPα и TPβ к TХA2);
• рецепторы из семейства иммуноглобулинов (GPVI, FcγRIIA);
• C-тип лектиновый рецептор (P-селектин);
• тирозинкиназные рецепторы (рецепторы к тромбопоэтину, Gas-6, эфрины и Eph киназы);
• смешанные типы рецепторов (CD63, CD36, лиганд 1 P-селектина, рецепторы фактора некроза опухоли — α-ФНО и пр.). Многие из них присутствуют и на других клетках, но некоторые экспрессируются только тромбоцитами.
Активация тромбоцитов сопряжена со структурными изменениями в цитоплазме, в которой имеются тубулярная система, немногочисленные митохондрии, гранулы гликогена, плотные и накопительные β- и α-гранулы, а также пероксисомы. Причем α-гранулы накапливают белки, обеспечивающие гемостатическую функцию тромбоцитов, например ФВ, фибриноген, P-селектин, PECAM-1, CD40 лиганд, (CD154), тромбоцитарный фактор-4, тромбоглобулин, тромбоспондин, PDGF, а также GPIIb/IIIa (αIIbβ3). В свою очередь β-гранулы богаты нуклеотидами (AДФ и АТФ), серотонином, гистамином, пирофосфатом и кальцием [18]. При активации происходит последовательное выделение содержимого гранул. Однако сам процесс тромбогенеза представляет собой сложный каскад реакций, реализуемых на разных уровнях, включая:
• внутриклеточный — проявляется экспрессией рецепторов, изменением формы за счет цитоскелета, секрецией гранул и адгезией тромбоцитов;
• межклеточный — обеспечивает взаимодействие между тромбоцитами, тромбоцитом и эндотелием, тромбоцитами и лейкоцитами, тромбоцитами и внеклеточным матриксом или факторами плазмы крови (см. таблицу);
Таблица. Рецепторы тромбоцитов, реализующих контактные взаимодействия
Примечание. GP — гликопротеин; ФВ — фактор Виллебранда.
• межтканевый — характеризуется взаимосвязью системы крови, сосудистого эндотелия и соединительной ткани.
Для образования тромба в месте повреждения стенки сосуда требуется координация последовательных реакций в пространстве и времени. К таковым относятся привлечение тромбоцитов к субэндотелию для создания монослоя активированных клеток (фаза инициации); рекрутирование и активация дополнительных тромбоцитов с сопутствующим освобождением агонистов тромбоцитов (фаза прогрессирования) и молекулярный каскад, предот в ращающий спонтанную дезагрегацию (фаза стабилизации) [7, 21, 29]. Существует также система негативной регуляции функции тромбоцитов, которая лимитирует неконтролируемый тромбоз. В норме поддерживается баланс между ингибирующими и активирующими системами в тромбоцитах.
Динамика тромбогенеза. Фаза инициации
Ключевым фактором, инициирующим тромбогенез, являются повреждение эндотелия сосудистой стенки и взаимодействие между тромбоцитами и компонентами внеклеточного матрикса, в частности ФВ, коллагеном, фибронектином, тромбоспондином и ламинином. На эти адгезивные взаимодействия во многом влияют реологические условия [30]. Так, при низкой скорости кровотока, например, в венах и крупных артериях адгезия тромбоцитов к сосудистой стенке определяется взаимодействием с фибриллярным коллагеном, фибронектином и ламинином. В условиях высокой скорости сдвига, например, в сосудах микроциркуляторного русла или в стенозированных артериях тромбоциты прикрепляются к субэндотелию (к ФВ) [7].
Растворимый ФВ не связывается с тромбоцитами, только форма, иммобилизированная на коллагене, является высоко реактивной в отношении тромбоцитов. Этот феномен связывают с изменением молекулы ФВ, поскольку в условиях натяжения домен А1 становится доступным для взаимодействия с тромбоцитами. Связывание тромбоцитов с ФВ является динамичным процессом, в котором начальная активация тромбоцитов характеризуется временным связыванием с участием комплекса GPIb—V—IX [29]. Эта реакция способствует дальнейшему формированию связей тромбоцитов, например, с коллагеном или с ФВ, но уже с помощью интегринов αIIbβ3. Известно, что комплекс GPIb—V—IX является главным тромбоцитарным рецептором, обеспечивающим взаимодействие с ФВ [16]. Этот комплекс состоит из гликопротеинов, богатых лейцином: GPIbα и GP Ibβ, которые связаны посредством дисульфидных связей и нековалентно — с GPIX (22 кД) и GPV в пропорции 2:4:2:1. У человека дисфункция этих рецепторов ассоциирована с развитием синдрома Bernard Soulier (ВSS) — генетически детерминированным кровотечением, развивающимся вследствие макротромбоцитопении и неспособности тромбоцитов формировать агрегаты в ответ на ристомицин [28]. Помимо ФВ комплекс GPIb—V—IX может связываться с другими адгезионными белками (коллагеном, тромбоспондином-1), α-тромбином и факторами коагуляции (кининоген, факторы XI и XII), а также обеспечивать взаимодействие с активированными эндотелиальными клетками и лейкоцитами за счет связывания с P-селектином и Mac-1 соответственно [8]. Ключевым компонентом комплекса GPIb—V—IX, как было установлено на генетически модифицированных мышах, является GPIbα. Именно его отсутствие сопровождалось нарушением тромбообразования, а использование моноклональных антител к GPIbα у обезьян вызывало значимое увеличение времени кровотечения [31]. Необходимо подчеркнуть, что дефицит ФВ может компенсироваться повышением связывания рецепторов с альтернативными лигандами.
Механизмы сигнализации при взаимодействии ФВ с GPIbα пока полностью не выяснены. Установлено, что цитоплазматический регион GPIbα связан с филамином (его называют также актинсвязывающим белком), кальмодулином и протеином 14-3-3z, а это предполагает возможность взаимодействия с такими сигнальными молекулами, как Ф3К, киназа фокальной адгезии (FAK), Src-связанная тирозинкиназа, ГТФаза-активирующие белки и тирозинфосфатазы (PTP1b и SHPTP10) [18]. Кроме того, комплекс GPIb—V—IX сопряжен с другими важными белками в регуляции функции тромбоцитов, включая GPVI, FcR γ-цепь, α2β1, FcγRIIA [28]. Предполагается, что наличие данных межмолекулярных связей возможно в специализированных доменах мембраны, известных как липидные щели, и имеет целью поддержание механизма активации тромбоцитов через сигнальный путь GPIbα [26, 29]. Таким образом, несмотря на отсутствие связи между GPIb—V—IX и тирозинкиназной активностью, данный комплекс не сопряжен напрямую с белками G и не содержит фосфорилированные остатки тирозина для прямой связи с сигнальными молекулами. Усиление сигнализации через GPIbα за счет иммобилизации ФВ вызывает активацию тромбоцитов, проявляющуюся типичным каскадом реакций — повышением внутриклеточного уровня Ca2+, фосфорилированием белков: ФЛСγ2, extracellularsignal regulated kinase ½ (ERK1/2), Syk, синтезом TХA2 с последующим освобождением AДФ и активацией αIIbβ3 [16]. Нужно отметить, что рецепторы αIIbβ3 были одним из первых типов рецепторов тромбоцитов, скрупулезно изученных и генотипированных. Это наиболее многочисленные рецепторы на поверхности тромбоцитов — их число может достигать '80 000 копий на одном тромбоците. Лигандами данных интегриновых рецепторов являются фибриноген и ФВ [8]. Механизм сигнализации через рецепторы этого типа будет описан ниже. В данном разделе мы лишь приведем факты о роли полиморфизма указанного типа рецепторов в патологии сердечно-сосудистой системы [19, 32]. Клинически значимыми считаются два наиболее важных аллеля b3, кодирующих Leu33 (PlA1 или HPA-1a) и Pro33 (PlA2 или HPA-1b), распространенность которых в европейской популяции составляет соответственно 0,85 и 0,15. Показано увеличение распространенности аллеля PlA2 (в 3,6 раза) у пациентов с инфарктом миокарда (ИМ) и нестабильной стенокардией в возрасте моложе 60 лет по сравнению с общей популяцией. В связи с этим сделано заключение о роли полиморфизма PlA2 как генетического фактора риска развития сосудистой патологии и ИМ [33].
Дополнительные исследования, направленные на анализ рецепторов к ФВ — GPIbα, позволили выявить ассоциацию между мутантным аллелем, кодирующим Met145 (более распространенным вариантом является Thr145), и риском развития коронарной патологии или приступа ишемии [23]. Другим вариантом диморфизма гена GPIbα является присутствие тимина или цитозина в позиции 25 от промотора кодона ATG [31]. Начальные клинические исследования не выявили связи между редко встречающимся вариантом 25C и риском развития острого коронарного синдрома. Однако позднее была выявлена ассоциация неблагоприятного исхода ИМ у пациентов с наличием 25C и Met145 [34].
Поскольку наиболее значимым элементом внеклеточного матрикса является коллаген, то изучение рецепторов к нему традиционно находится в центре исследований, посвященных механизмам нарушения тромбогенеза. Как и ФВ, коллаген является важнейшим инициатором адгезии тромбоцитов [27]. В сосудистой стенке выявлены коллагены I, III, IV, VI, XVIII типов. Коллагены I, III и VI типов имеют сродство к ФВ и молекулам, ассоциированным с внеклеточным матриксом. Механизмы, с помощью которых коллаген вызывает адгезию и активацию тромбоцитов, во многом остаются неизученными. На поверхности тромбоцитов описано два типа рецепторов к коллагену — это представитель семейства иммуноглобулинов GPVI и интегрин α2β1. Причем GPVI (62 кД) является молекулой с низким сродством к коллагену, но с высокой способностью к активации тромбоцитов [18]. Данный рецептор представлен двумя внеклеточными участками иммуноглобулинов, имеет муциноподобный стержень, короткий связующий пептидный фрагмент, трансмембранный домен и короткий цитоплазматический хвост. Оказалось, что GPVI конститутивно формирует комплексы (димеры) с γ-цепью Fc-рецепторов, которые относятся к иммунорецепторам [19]. Последние ассоциированы с ITAM, являющимся субъединицой рецептора, передающей сигнал. Когда рецепторы GPVI связываются поперечными связями при взаимодействии с коллагеном или специфическими лигандами (такими как convulxin или alborhagin), Src-киназы фосфорилируют последовательность ITAM в цепи FcRγ, вызывая сборку и активацию Syk с последующей активацией нисходящего сигнального пути.
При этом формируются сигналосомы, включающие различные адаптерные и эффекторные белки (LAT, SLP-76, Gads), которые индуцируют активацию ФЛСγ2, что ведет к освобождению ДАГ и И3Ф, запускающих активацию тромбоцитов [18]. У мышей с утратой GPVI или FcRγ-цепи существенно нарушен ответ тромбоцитов на коллаген и ингибируется процесс образования тромба. Кроме того, использование фрагмента Fab — новых моноклональных антител к GPVI человека, вызывает ингибирование тромбоза без увеличения времени кровотечения, что принципиально отличается от эффекта антител против GPIIb/IIIa.
Вторым рецептором, играющим важную роль в адгезии тромбоцитов к коллагену, является интегрин α2β1, который относится к GPIa/IIa [28]. Уровень экспрессии α2β1, как и GPVI, характеризуется полиморфизмом, степень которого коррелирует со скоростью адгезии тромбоцитов и выраженностью ответа на коллаген [30]. Роль рецепторов α2β1 в гемостазе подтверждается склонностью к умеренным кровотечениям и нарушением агрегации тромбоцитов на коллаген у субъектов с врожденным снижением экспрессии этого интегрина. У мышей с дефицитом α2 или b1 зарегистрировано нормальное время кровотечения, но отмечается снижение адгезии и агрегации тромбоцитов в ответ на коллаген. Конформационная активация α2β1 повышает их сродство к коллагену и последующий каскад реакций, сопровождающийся увеличением количества GPVI или активацией αIIbβ3. Кроме того, связывание коллагена с α2β1 также является триггером в сигнализации, индуцируемой GPVI, что усиливает активацию тромбоцитов. Дефицит α2β1 и GPVI у мышей вызывает полное ингибирование образования тромба, т.е. для оптимальной активации тромбоцитов необходим синергизм эффектов α2β1 и GPVI [29]. Данное положение было подтверждено и в исследованиях на тромбоцитах человека. Показано, что у здоровых доноров содержание GPVI во многом соответствует плотности α2β1. Этот факт объясняется взаимодействиями между рецепторами данных типов, которые функционально сопряжены друг с другом на поверхности тромбоцитов. Предполагается, что и на генетическом уровне экспрессия одного типа рецептора сопряжена с экспрессией и плотностью другого типа во время мегакариоцитопоэза, хотя в ряде работ показана независимая транскрипционная регуляция экспрессии обоих генов [8]. В другой работе [23] продемонстрированы не только координация экспрессии GPVI и α2β1 при дифференцировке мегакариоцитов, но и сопряженность с экспрессией CD41. Ввиду данного параллелизма особого интереса заслуживает вопрос о том, полиморфизм какого типа рецепторов ассоциирован с нарушением реакции тромбоцитов на коллаген и повышением риска сердечно-сосудистой патологии.
Связь лиганда с рецептором GPVI, при его коэкспрессиии с γ-цепью Fc-рецептора, ведет к запуску сигнальных реакций и к активации интегриновых рецепторов α2β1, усиливающих сигнальные реакции внутри тромбоцита. После отложения монослоя тромбоцитов на подложке ФВ и коллагена включается следующий этап, направленный на привлечение дополнительных тромбоцитов — агрегация. В ответ на инициацию тромбогенеза со стороны коллагена или ФВ происходит стимуляция зависимого от циклооксигеназы (ЦОГ) образования TХA2 и секреция гранул, содержащих АДФ. Этот шаг делает возможным локальное накопление растворимых агонистов. Финальным шагом является активация αIIbβ3, вызывающая конформационные изменения, что обеспечивает возможность связи с фибриногеном и ФВ, формирующих стабильные связи между тромбоцитами. Огромное количество αIIbβ3 копий на поверхности тромбоцитов (40 000—80 000) позволяет формировать крупные агрегаты в месте повреждения сосудистой стенки [2, 7].
Фаза прогрессирования
Ключевыми стимуляторами фазы прогрессирования являются гуморальные (в плазме крови) и аккумулированные в плотных гранулах тромбоцитов факторы AДФ, TХA2, адреналин, серотонин и тромбин. Описано 2 основных вида АДФ — экзогенный, освобождаемый при повреждении из эритроцитов, и эндогенный — секретируемый тромбоцитами при индукции коллагеном и ФВ [28]. Действие гуморальных регуляторов фазы прогрессирования опосредовано активацией рецепторов, связанных с белком G, которые представлены несколькими семействами белков, связывающих гуаниннуклеотид: Gq, Gi, Gs, Gz, G12, G13 [35]. Именно они играют ключевую роль в образовании тромба при действии растворимых агонистов, активируя ассоциированные эффекторы: аденилатциклазу (АЦ), ФЛC, Ф3К, p115-RhoGEF. Суммация эффектов стимуляции разных рецепторов, связанных с белком G, сопровождается комплексом реакций, включая следующие:
• стимуляция изоформы ФЛCb через Gq, что приводит к повышению уровня цитозольного Ca2+ и активации протеинкиназы C (ПкС);
• реорганизация актинового цитоскелета через G12 и G13, что вызывает изменения архитектоники кольца микротрубочек, формирование филоподий и ламеллоподий;
• снижение активности АЦ и образование цAMФ через семейство белков Gi [2]. Повышение уровня цАМФ возможно за счет действия на тромбоциты факторов эндотелиального происхождения — простациклина и оксида азота (NO).
Исследования на животных с генетическим дефектом экспрессии отдельных белков G помогли определить их роль в активации тромбоцитов и формировании тромба. Для понимания механизмов данной суммации необходимо рассмотреть набор рецепторов, активируемых разными растворимыми агонистами.
На тромбоцитах представлено три варианта пуриновых рецепторов: Р2Х1 — катионный канал, активируемый АТФ; и два варианта рецепторов, ассоциированных с белками G — P2Y1 и P2Y12, активируемых АДФ [27]. Для физиологической агрегации, индуцированной АДФ, необходима совместная активация рецепторов P2Y1 и P2Y12. Селективная активация рецепторов Р2Y1 вызывает умеренную временную и нестабильную агрегацию тромбоцитов. Данный тип рецепторов ассоциирован с Gq, вызывает мобилизацию Са2+ из внутриклеточных депо, что ведет к изменению формы тромбоцитов и временной агрегации в ответ на АДФ [26]. Фармакологическое ингибирование или генетический дефицит рецепторов Р2Y1 обусловливает отсутствие ответа тромбоцитов на АДФ. На внутриклеточном уровне это сопровождается нарушением Са2+-сигнализации, хотя при этом способность АДФ ингибировать уровень внутриклеточного цАМФ сохраняется [36]. Рецепторы P2Y1 также участвуют в агрегационном ответе на коллаген и играют ключевую роль в индуцированном коллагеном изменении формы даже в условиях ингибирования ЦОГ и продукции ТХА2. Рецепторы P2Y1 обеспечивают активацию p160 Rho-киназы, что вместе с повышением уровня Са2+ определяет изменение формы тромбоцитов при действии AДФ [37]. Изучение роли рецепторов P2Y1 в контроле агрегации тромбоцитов и тромбоза продемонстрировало высокую эффективность блокады данных рецепторов. Причем ингибирование рецепторов P2Y1 вызывает умеренное увеличение времени кровотечения. У P2Y1-дефицитных мышей и у животных, получавших селективный антагонист P2Y1, выявлена резистентность к системной тромбоэмболии, вызванной инфузией смеси коллагена и адреналина, или тромбоэмболии, связанной с инъекцией тканевого фактора. Подтверждено участие рецепторов P2Y1 в активации экспозиции P-селектина, формировании тромбоцитарно-моноцитарных агрегатов и экспрессии тканевого фактора при стимуляции тромбоцитов АДФ, коллагеном или низкими концентрациями агонистов тромбиновых рецепторов [26]. Предполагается также важная патогенетическая роль рецепторов P2Y1 в атеротромбозе, поскольку у P2Y1-дефицитных мышей, скрещенных с животными, имеющими дефект гена аполипопротеина E (ApoE–), наблюдали снижение размера атеросклеротических бляшек [16].
Проведенные фармакологические исследования показали, что сами рецепторы P2Y1 не способны воспроизвести на тромбоцитах все отмеченные выше эффекты АДФ, и ряд параметров зависит от участия рецепторов P2Y12. Активация рецепторов Р2Y12 под действием АДФ сопровождается активацией белка Gi и играет центральную роль в амплификации активации тромбоцитов в ответ на другие более мощные агонисты (тромбин, коллаген, тромбоксан А2). Внутриклеточный путь, через который рецепторы P2Y12 усиливают ответ тромбоцитов, включает угнетение продукции цАМФ за счет ингибирования АЦ с последующим ограничением активности протеинкиназы А, дефосфорилирование фосфопротеина, стимулированного вазодилатацией, и малых ГТФаз (Rap1B) [28]. Важную роль в реализации ответа тромбоцитов на АДФ играет опосредованная рецепторами P2Y12 активация Ф3К, которая, модулируя рецепторы И3Ф, усиливает открытие кальциевых каналов в ЭС тромбоцитов и выход Са2+. Ингибирование рецепторов Р2Y12 может ослаблять агрегацию тромбоцитов, а также их прокоагулянтную и провоспалительную активацию (что отмечается при коронарном атеросклерозе) [1, 38].
Анализ обсуждаемой проблемы порождает ряд вопросов: почему AДФ является слабым агонистом по сравнению, например, с TXA2 или тромбином? Почему АДФ индуцирует только обратимую агрегацию и не может самостоятельно вызывать секрецию гранул тромбоцитов? С нашей точки зрения, этот феномен обусловлен очень низким уровнем экспрессии рецепторов P2Y на плазмолемме тромбоцитов по сравнению с другими рецепторами, связанными с белком G. На тромбоцитах представлено около 150 участков, связывающих рецептор P2Y, что очень мало по сравнению с плотностью рецепторов к тканевому фактору или тромбину (РAR-1) (соответственно от 1000 до 2000 мест на тромбоците) [2, 39].
Не менее важным и клинически значимым является вопрос о сопряженности эффектов АДФ и тромбоксана в прогрессировании тромбогенеза. Повышение продукции ТХА2 считается одним из ключевых механизмов развития тромботических заболеваний, включая ИМ, нестабильную стенокардию, эмболию легочной артерии, атеросклероз. В тромбоцитах ТХА2 вызывает изменение формы, гидролиз фосфоинозитида, мобилизацию Са2+, фосфорилирование белков, секрецию гранул и агрегацию тромбоцитов [40]. Синтезированный ТХА2 диффундирует через мембрану тромбоцитов и активирует рекрутирование других тромбоцитов, вызывая рост тромба. Рецепторы и ферменты синтеза АДФ и ТХА2 являются мишенями фармакологической коррекции при различных заболеваниях сердечно-сосудистой системы [41—43]. Более того, в клинике отмечено частое сочетание резистентности к тиенопиридинам (блокаторам Р2Y12 рецепторов) и ацетилсалициловой кислоте, ингибирующей ЦОГ и образование тромбоксана А2 [43, 44]. Нужно отметить, что многие рецепторы и трансдукторы вовлечены в регуляцию активности фосфолипазы A2(ФЛА2), а следовательно, и образования ТХА2, включая ПкC, MAPKs и Са2+ [2]. Ключевым фактором сопряжения при этом является процесс фосфорилирования ФЛА2, в котором доказана важная роль ряда киназ — ERK, Srk, p38-MAPK.
Действительно, активация P2Y1 и P2Y12 сопровождается усилением синтеза ТХА2. Важную роль в АДФ-ассоциированной продукции ТХА2 играют ERK1/2, которые относятся к семейству тромбоцитарных MAP-киназ и активируются под действием различных агонистов, наиболее известными среди которых являются тромбин и конволюксин [7]. AДФ активирует ERK2 при стимуляции рецепторов P2Y за счет механизма, сопряженного с активацией семейства киназ Src. При этом требуется активация обоих типов рецепторов P2Y1 и P2Y12 [36]. Активация ERK2 возможна при стимуляции рецепторов P2X1, что, вероятно, связано с повышением входа Са2+, тогда как внеклеточный Са2+ негативно регулирует генерацию ТХА2 тромбоцитами. Следовательно, снижение уровня внеклеточного Са2+ ведет к повышению стимулированного агонистами синтеза ТХА2. Закономерно, что ингибиторы сигнальных событий, ведущих к активации Gq, ФЛCβ и кальция, блокируют как фосфорилирование ERK2, так и генерацию тромбоксана [6]. Ингибирование всех изоформ ПкC потенцирует фосфорилирование ERK2, что отражает негативную роль ПкC в регуляции пути ERK. Известна аналогичная регуляция пути Gq изоформами ПкC. Например, ПкCα ингибирует активность ФЛCβ1 [28], а ингибиторы ПкC вызывают значительное повышение продукции тромбоксана.
TХA2 является вазоконстриктором и мощным агонистом тромбоцитов, вызывая изменение их формы, гидролиз фосфоинозитида, мобилизацию Са2+, фосфорилирование белков, секрецию и агрегацию [33]. Синтезированный ТХА2 диффундирует через мембрану тромбоцитов и активирует рекрутирование других тромбоцитов, вызывая рост тромба. Рецепторы к TХA2 представлены в 2 вариантах — TPα и TPβ, которые отличаются только по C-концевому цитоплазматическому домену и кодируются одним геном на 19p13.3 [32]. Рецепторы к ТХА2 ассоциированы с белками G и отличаются паттерном сигнализации. Рецепторы к тромбоксану А2 на плазмолемме тромбоцитов сопряжены с Gq и G13 активаторами ферментов, но не с белком Gi. Стимуляция Gq вызывает усиление образования И3Ф и мобилизацию Са2+, т. е. запускает реакции, развивающиеся при активации P2Y1; эффект стимуляции G12/13 вызывает активацию Rho ГТФаз и изменение формы тромбоцитов. Совместная блокада Gq и G 13 характеризуется выключением ответа на TXA2 [2].
Среди всех стимуляторов прогрессирования тромбогенеза наиболее эффективным считается тромбин, который вызывает полный каскад реакций, включая изменение формы тромбоцитов, секрецию, генерацию ТХА2, мобилизацию Са2+ и агрегацию. Для образования тромбина требуется включение ряда реакций на поверхности тромбоцита, ассоциированных с теназным комплексом (фактор IXa в комплексе с фактором VIIa, активирующим X) и протромбиназным комплексом (фактор Xa в комплексе с Va, который активирует II) [45]. Тромбин может активировать тромбоциты в очень низкой концентрации (0,1 нМ), при этом мощно стимулируется ФЛCb, повышая внутриклеточный уровень Са2+ в течение нескольких секунд, что вызывает последующую активацию ФЛА2. Вызванный тромбином ответ отчасти опосредуется через комплекс GPIb/IX/V, но главным образом за счет PAR-1 и PAR-4 у человека, и PAR-3 и PAR-4 у мыши. Субъединица комплекса GPIbα GPIb/IX/V содержит сайт с высоким сродством к α-тромбину [7]. Связывание α-тромбина с GPIbα индуцирует адгезию тромбоцитов, их прогрессивное накопление, секрецию биологически активных веществ (БАВ) и агрегацию тромбоцитов. Блокада взаимодействия тромбина и GPIbα с помощью антител вызывает слущивание с поверхности PAR-1, на основании чего был сделан вывод, что GPIbα может быть кофактором, который способствует доставке тромбина к поверхности тромбоцита, где тромбин оказывает протеолитическое действие на свои рецепторы [39].
Несмотря на то что клонирование рецепторов к тромбину (т. е. PARs) было выполнено еще в 1991 г., а за последующие десятилетия выяснены молекулярная структура, механизмы регуляции и особенности экспрессии PAR в организме человека, в отношении семейства данных рецепторов больше вопросов, чем ответов [7, 28, 39]. И это при том, что процесс тромбообразования и сопряжение между системой коагуляции, воспалением, ангиогенезом и репарацией сосудистой стенки и периваскулярного региона невозможны без участи PARs. Многообразие эффектов при активации PAR-1 связано с участием нескольких разновидностей белков G, ассоциированных с разными киназами. Реализация сигнала при активации PAR-1 связана с участием белка G, ФЛС, ДАГ, И3Ф, протеинкиназы В, ПкС, МАРК, р42/44 [16], что побуждает к поиску ответа на вопрос: от чего зависит включение того или иного сигнального пути при активации PAR-1? Ключевым процессом в реализации проагрегантного эффекта PAR-1 является мобилизация Са2+. Взаимосвязь тромбина с PAR-1 ведет к изменению формы тромбоцитов и сопровождается освобождением серотонина (5-НТ), АТФ, ТХА2 и других компонентов гранул. Не менее важным эффектом тромбина на тромбоциты является транслокация P-селектина и лиганда CD40 на плазматическую мембрану, что усиливает связь между тромбоцитами и эндотелием [29]. Описано также усиление образования других факторов, включая VEGF, что запускает инициацию ангиогенеза. Системная роль PAR-1 в этом процессе подтверждается экспериментами на трансгенной линии мышей PAR–/PAR–. При формировании данной линии отмечена гибель эмбрионов на ранних стадиях развития вследствие блокады ангиогенеза [23].
PAR-1 и нисходящий поток сигнализации при действии тромбина необходимы в эндотелии для регуляции ангиогенеза и развития сосудов. У мышей с PAR-4–/– отмечено отсутствие ответа даже на высокие концентрации тромбина. Предполагается, что у человека PAR вовлечены в патогенез атеротромбоза [16]. Установлено, что PAR-1 вызывает активацию эндотелиальных клеток за счет усиления освобождения ФВ и поверхностной экспрессии P-селектина, усиливая ролинг и адгезию тромбоцитов и лейкоцитов. Тромбин также вызывает усиление продукции БАВ, в том числе фактора активации тромбоцитов, простагландинов, хемокинов и пр., разными клетками, стимулируя воспаление. Поэтому в настоящее время PARs рассматриваются как потенциальные мишени антитромбоцитарной терапии.
И наконец, завершая рассмотрение роли тромбина в регуляции образования тромба, необходимо подчеркнуть, что, в отличие от АФД и ТХА2, вовлекаемых в гемостаз как в норме, так и при патологическом тромбозе, PAR-1 индуцирует активацию тромбоцитов только в условиях формирования окклюзионного тромба, богатого тромбоцитами, но не принимает участия в защитном гемостазе и формировании начального монослоя тромбоцитов в зоне повреждения. В ряде исследований показано, что опосредованное тромбином расщепление фибриногена до фибрина является более важным механизмом гемостаза, чем опосредованная тромбином активация тромбоцитов. У мышей с дефицитом фибриногена или PAR-4 наблюдались мощные профузные кровотечения, снижение количества и нарушение активации тромбоцитов, тогда как дефицит или блокирование РАR-1 не сопровождались изменением количества и формы тромбоцитов, развитием спонтанных кровотечений. Эти исследования привели к разработке нового класса антитромбоцитарных препаратов — ингибиторов PAR-1, одним из селективных антагонистов которого является FR171113 [44]. Его апробация в эксперименте продемонстрировала снижение вероятности и выраженности артериального тромбоза без увеличения времени кровотечения и нарушения параметров коагуляционной системы крови. Данный класс препаратов имеет определенные перспективы направленной коррекции гемостаза при патологии сердечно-сосудистой системы.
В регуляции тромбогенеза важным является стимулирующий эффект тромбина на транслокацию в мембрану интегрина aIIbb3, связывающего фибриноген и ФВ, что ведет к мощной агрегации тромбоцитов [26]. Для активации интегрина αIIbβ3 требуется инициация всего спектра перечисленных выше реакций, включая активацию одной или нескольких изоформ ФЛC, повышение уровня внутриклеточного Ca2+, стимуляцию ПкС и Ф3K, вызывающих реорганизацию цитоскелета тромбоцитов и активацию его белков, включая талин [26, 37]. Активированный талин может связываться с цитоплазматическим доменом субъединицы α3 интегрина, вызывая диссоциацию цитоплазматического хвоста и трансмембранного домена αIIb и β 3, что способствует олигомеризации интегринов и связыванию фибриногена. Модуляция сродства αIIbβ3 возможна через сигнальный комплекс Rap-1—RIAM—талин [2]. Триггером активации β-интегрина и связи его с талином является белок киндлин-3. Дефект экспрессии интегринов лежит в основе развития синдрома «ленивых лейкоцитов» и кровотечений по типу Гланцмана. Эти заболевания могут быть связаны с дефектом активности или экспрессии Rap-1 или киндлин-3. В активации интегриновых рецепторов αIIbβ3 также принимает участие Аkt-1 (протеинкиназа В). Данный трансдуктор также обеспечивает реализацию сигнала, запускаемого тромбином и коллагеном [46].
И наконец, недавно выявлен новый механизм влияния тромбина на активность тромбоцитов с участием металлопротеиназ, активируемых через PAR-1. Обнаружено, что экспозиция тромбоцитов с коллагеном вызывает активацию ММР-1, которая в свою очередь отщепляет N-концевой внеклеточный домен PAR-1, независимо от тромбина [47]. Трансформируемый MMP-1 рецептор или растворимый аналог пептида является мощным стимулятором G12/13-Rho-зависимого сигнального пути, вызывая хемотаксис и МАРК-каскад в тромбоцитах и других клетках [48]. Это отщепление также генерирует длинный пептидный лиганд, который может быть агонистом, связывающимся с PAR-1. Блок пути MMP-1— PAR-1 ведет к ингибированию зависимого от коллагена тромбогенеза, предотвращает артериальный тромбоз и ретракцию тромба, что предполагает возможность развития новой тактики лечения при коронарном синдроме, мишенью которой может стать металлопротеазо-рецепторная система [49].
Фаза стабилизации
Стабилизация тромба реализуется за счет каскада сигнальных реакций, инициируемых формированием контактов между тромбоцитами в процессе агрегации. При этом между тромбоцитами образуются прочные контакты с щелью шириной менее 50 нм за счет формирования прямых и непрямых мостиков, что определяет возможность паракринной регуляции соседних тромбоцитов [18]. Эти узкие контакты также ограничивают диффузию плазменных факторов, предотвращая преждевременное фибринолитическое действие плазмина в области растущего тромба.
Фаза стабилизации характеризуется включением контакт-зависимой системы сигнализации с участием интегринов, в частности αIIbβ3 [2]. Лигандом αIIbβ3 является преимущественно фибриноген, играющий важную роль в формировании тромба за счет реорганизации цитоскелета, формирования агрегатов, прокоагулянтной поверхности и ретракции тромба, что обеспечивает сужение щелей между тромбоцитами и позволяет увеличить локальную концентрацию агонистов тромбоцитов. αIIbβ3 рассматривается как терапевтическая мишень антитромбоцитарной терапии у пациентов, подвергающихся чрескожным коронарным вмешательствам, и при лечении нестабильной стенокардии [5, 50].
Активация αIIbβ3 включает помимо механизмов «изнутри наружу» сигнальный путь, связанный с тирозиновым фосфорилированием и формированием крупного сигнального комплекса между цитоплазматическим доменом αIIbβ3 и внутриклеточными белками, включая FAK, талин, миозин, 3-эндонексин, CIB1, Shc, Src и Syk, PRP-1b-тирозинфосфатазу и ПKCα [7]. Роль αIIbβ3 сигнализации «снаружи внутрь» (outside—in) в усилении агрегации тромбоцитов продемонстрирована на линии трансгенных мышей, у которых была замена (мутация) остатков Tyr-747 и Tyr-759 на фенилаланин. Данная линия мышей характеризовалась нарушением сигнализации «снаружи внутрь», что сопровождалось формированием нестабильных агрегатов [28]. Специфические механизмы связей внутритромбоцитарных белков с αIIbβ3 и их физиологическое участие в сигнальных путях, запускаемых интегринами, остаются неясными и активно изучаются.
Однако не только интегрины являются участниками формирования тромба. Важную роль в стабилизации тромба играют соединяющие молекулы адгезии (junctional adhesion molecules — JAM-A и JAM-C), которые поддерживают контакты и сигнализацию между тромбоцитами, а также тромбоцитами и лейкоцитами. Аналогична роль SLAM (CD150), члена CD2 семейства молекул адгезии, которые экспрессируются в тромбоцитах и подвергаются тирозиновому фосфорилированию при формировании агрегатов. Исследование на мышах SLAM–/– показало нарушение ответа тромбоцитов на коллаген и при стимуляции PAR-4 [32]. На поверхности активированных тромбоцитов присутствует лиганд CD40 (CD40L, CD154), белок семейства α-ФНО, принимающий участие в тромбогенезе. При формировании тромба происходит прогрессирующее слущивание лиганда с поверхности тромбоцитов с образованием растворимой формы sCD40L, которая может связываться со своим рецептором CD40, а также с αIIbβ3, модулируя внутриклеточную сигнализацию через интегрины. Дефицит CD40L снижает стабильность тромба при повреждении сосудов [51].
Иной механизм сигнализации в тромбоцитах запускается при активации эфринами семейства Eph рецепторных тирозинкиназ. Тромбоциты человека экспрессируют EphA4, EphB1 и эфрин В, их формирование сопряжено с экспозицией αIIbβ3 в активированных тромбоцитах. Кластеризация EphA4 или эфрина B1 вызывает адгезию тромбоцитов к иммобилизированному фибриногену. Блокада комплекса Eph—эфрин вызывает уменьшение тромбоза за счет нарушения b3-фосфорилирования, ингибирования агрегации тромбоцитов и нарушения реакции на коллаген [7]. В литературе описаны еще два дополнительных лиганд-рецепторных взаимодействия, направленных на формирование близких и прочных контактов между тромбоцитами. Это связи семафорина 4D (Sema4D) и Gas-6 с их тромбоцитарными рецепторами. Sema4D (CD100) относится к I типу мембранных гликопротеинов, роль которых была описана ранее при активации Т-лимфоцитов. Связывание Sema4D с рецепторами, CD72 и плексином-B1 регулирует образование тромба. У мышей Sema4D–/– нарушен ответ тромбоцитов на коллаген, но сохраняется нормальная реакция на AДФ и PAR-4; уменьшен размер тромбов при повреждении стенки сосудов [25]. Фрагменты Sema4D могут стимулировать репарацию эндотелия. Коррекция молекул Gas-6, продукта гена 6, также считается перспективным направлением коррекции гомеостаза сосудистой стенки, поскольку останавливает рост клеток. Это витамин К-зависимый белок, вовлеченный в регуляцию роста, адгезии, миграции клеток при взаимодействии с Axl, Tyro 3 и Mer тирозинкиназными рецепторами. У мышей Gas-6 обнаружен в плазме и α-гранулах тромбоцитов. Дефицит Gas-6 или их рецепторов ведет к ограничению ответа тромбоцитов на агонисты и препятствует тромбообразованию. Реализация эффектов Gas-6 обусловлена реверсией aIIbb3 сигнала «снаружи внутрь» за счет активации Ф3К и Akt, что вызывает фосфорилирование субъединицы интегрина β3 и последующую ретракцию тромба [28]. Возможности ингибирования сигнализации Gas-6 исследуются в плане разработки новых антитромбоцитарных препаратов.
Негативная регуляция активации тромбоцитов и роста тромба
Формирование тромба является динамичным процессом, в котором ограничение размеров тромба контролируется NO и простациклином. Активация тромбоцитов может блокироваться на уровне сигнальных путей через молекулы адгезии PECAM-1 (известные также как CD31). Установлено, что PECAM-1 участвует в поддержании формирования тромба путем вовлечения GPVI, GPIb, тромбина с его рецепторами, а также αIIbβ3 [52]. Подобно GPVI PECAM-1 является представителем суперсемейства иммуноглобулинов, имеет 6 внеклеточных и 1 трансмембранный домен, а также цитоплазматический хвост [34]. В последнем имеется содержащий тирозин ингибиторный фрагмент (ITIM), который фосфорилируется при стимуляции гомофильных взаимодействий и/или кластеризации, усиливая включение тирозина, серина/треонина и, возможно, липидных фосфатаз, что вызывает последующее ингибирование киназозависимой сигнализации. Подобно PECAM-1 еще одна молекула на поверхности тромбоцитов — ESAM принимает участие в росте тромба [27].
Активация αIIbβ3 и, вероятно, других интегринов не является необратимой. Это динамичный двунаправленный процесс, обеспечивающий формирование прокоагулянтной поверхности тромбоцитов с пролонгированным поддержанием высокого уровня Са2+. В этих условиях поддержание функциональной активности тромбоцитов зависит от удаления рецепторов с их поверхности; процесс опосредуется металлопротеиназами из семейства дезинтегринов (ADAM) [53]. Привлекательным механизмом ограничения формирования тромба у больных представляется целенаправленное «слущивание» с поверхности тромбоцитов эктодоменов адгезионно-сигнальных рецепторов — GPIba и GPVI или отщепление с помощью ADAM-13 ФВ, накапливаемого на поверхности тромба [2, 48].
Важную роль в контроле гемостаза играет механизм десенситизации рецепторов, обеспечивающих адаптацию клеток на действие стимулов. Так, установлено, что при начальной индуцируемой АДФ активации тромбоциты временно не отвечают на повторные стимулы агониста. Аналогичные процессы описаны для адренорецепторов и рецепторов к серотонину [54]. В основе данного феномена лежит механизм интернализации рецепторов. Рассматриваются прикладные аспекты блока механизмов димеризации гликопротеиновых и иммуноглобулиновых рецепторов, часто формирующих гомо- и гетеродимеры.
Таким образом, тромбогенез представляет собой сложный многоэтапный и многофакторный процесс, в котором происходит взаимодействие компонентов сосудистой стенки, тромбоцитов, факторов коагуляции, гуморальных (растворимых) регуляторов. Ответ тромбоцитов в разные фазы тромбогенеза зависит не только от лиганд-рецепторных взаимодействий, но и во многом определяется суммацией позитивных и негативных модуляторов тромбогенеза на уровне систем внутриклеточной сигнализации. В конечном итоге ключевым звеном ответа тромбоцитов является уровень внутриклеточного Са2+, однако сопряженная стимуляция рецепторных и нерецепторных тирозиновых киназ, серин-треониновых киназ и фосфатаз определяет включение механизмов прогрессирования и стабилизации образования тромба, усиление прокоагулянтной и провоспалительной активности тромбоцитов, их участие в регуляции репарации сосудистой стенки и ремоделирования тканей и органов. Понимание молекулярных механизмов функционирования тромбоцитов позволит оптимизировать стратегию управления тромбогенезом и сопряженным с ним воспалением как в условиях его усиления при атеротромбозе, так и в условиях нарушения при кровотечениях разного генеза.



{kind=link}
{kind=link}