
В России, как и в других экономически развитых странах, ведущей причиной смерти являются болезни системы кровообращения. Однако в нашей стране смертность от болезней сердца и сосудов более чем в 3 раза превышает таковую в странах Западной Европы и США, причем россияне умирают от этих причин в существенно более молодом возрасте [1]. В свою очередь, одной из основных причин смерти от сердечно-сосудистых заболеваний — (ССЗ) (около 50%) являются нарушения ритма и проводимости сердца [2]. Постоянное накопление информации о патогенезе ССЗ привело к пониманию, насколько значительную роль в их развитии играют генетические факторы. Практически не осталось болезней, в формировании которых не было бы установлено наследственной компоненты.
Синдром слабости синусового узла (СССУ) — сочетание клинических, электрокардиографических и электрофизиологических признаков, отражающих структурные повреждения синусового узла (СУ), его неспособность нормально выполнять функцию водителя ритма сердца и обеспечивать регулярное проведение автоматических импульсов к предсердиям. Наследственный СССУ возникает из-за уменьшения числа специализированных клеток в СУ, пролиферации соединительной ткани из-за различных генетических мутаций [3]. В 2003 г. D.W. Benson и соавт. представили мутантные гены СССУ. Это патология гена натриевого канала сердца (SCN5A) с выявленными 3 видами мутаций: 4 миссенс-мутации (T2201, P1298L, G1408R, R1632H), внутрирамочная делеция (del F1617), нонсенс-мутация (R1623X) и патология гена HCN4, локализованного на 15-й хромосоме при наследственном СССУ [4]. Исследования E. Schulz-Bahr и соавт. в этом же году подтвердили, что СССУ может возникать при патологии того же гена, и выявили, что нарушение работы ионных каналов СУ приводит к замедлению скорости спонтанной деполяризации и уменьшению частоты синусового ритма [5]. R. Milanezi и соавт. в 2006 г. скринировали 52 случая брадикардии на мутацию в области пейсмекерного канала гена HCN4 (Ser672Arg) [6]. F. Kyndt и соавт. (2001) описали мутацию GLY1408ARG в гене SCN5A в большой французской семье с СССУ и синдромом Бругада, передающимися по аутосомнодоминантному типу [7].
Однако все перечисленные гены не имеют мажорных мутаций, которые легко детектировать, что требует полного секвенирования этих генов. В любом случае это не отменяет необходимости исследования полиморфизма генов-модификаторов, белковые продукты которых участвуют в патогенезе заболевания. СССУ, обусловленный мутациями в генах, которые регулируют функционирование клеток СУ и сино-атриального проведения, практически не изучен. В 2009 г. нами опубликованы данные о том, что гетерозиготный генотип гена β1-адренорецепторов Ser49Gly и гомозиготный полиморфизм гена транскрипционного фактора SP4 Т80807Т являются генетическими предикторами данного синдрома. В настоящее время мы проводим расширение базы данных больных с СССУ и поиск новых генетических предикторов этого заболевания.
Ген α2β-адренорецептора (ADRA2B) расположен на 2-й хромосоме, не имеет интронов. Он кодирует α2β-адренергический рецептор [8], который является членом семейства α2-адренергических рецепторов, к которому помимо него относятся также субтипы α2А и α2С [9]. Впервые I/D-полиморфизм гена ADRA2B был описан в 1999 г. Делеция имеет небольшой размер (9 п.н.) и приводит к исчезновению 3 глутаминовых кислот в 3-й внеклеточной петле белка, что существенно влияет на функционирование рецептора. Ген ADRA2B имеет функциональный делеционный полиморфизм Ins>Del Codon 299. Исследование 380 здоровых японцев показало, что I/D-полиморфизм гена ADRA2B является достаточно распространенным в данной этнической группе [10]. Распространенность редкого аллеля составляет 35%. Частота сердечных сокращений у гомозигот DD была значительно снижена по сравнению с таковой при других генотипах. Поэтому представляет интерес выяснение распространенности этого аллеля у больных с СССУ и их родственников по сравнению с контрольной группой.
Одним из генов-кандидатов СССУ является ген eNOS. Он регулирует кальциевые L-каналы, обеспечивающие нормальный синусовый ритм и сокращение кардиомиоцитов. eNOS находят в нервных волокнах, иннервирующих синоатриальный и атриовентрикулярный узлы. Уменьшение NO приводит к возникновению номотопных аритмий в результате увеличения кальциевого потока. Влияние NO на ритм сердца обусловлено также его центральными эффектами. Помимо того, NO угнетает пролиферацию гладких мышечных клеток и может выступать в роли антиростового фактора, препятствующего пролиферации гладких мышечных клеток стенки сосудов и клеток СУ [11].
Эндотелиальная NO-синтаза — один из наиболее значимых источников физиологической связи NО и сердечно-сосудистой системы. Эта изоформа присутствует в эндотелиальных клетках сосудов и ответственна за вызываемую NO вазодилатацию, ингибирование атеросклероза и предупреждение тромбоза, а также внезапную сердечную смерть [12].
Ген, кодирующий eNOS, находится на хромосоме 7q35—36, состоит из 26 экзонов и кодирует белок молекулярной массой 135 кД, состоящий из 1203 аминокислот. Промотор гена eNOS содержит несколько доменов, т.е. может регулироваться рядом факторов транскрипции. Наиболее мощным регулятором экспрессии eNOS является напряжение сдвига, связанное с воздействием потока крови на поверхность эндотелиальных клеток [13].
Наиболее значимые 4 полиморфных маркера гена eNOS: интрон 18-й локус А27С; интрон 23-й локус G10T; интрон 4 eNOS 4а/b полиморфизм и аксон 7 Glu298Asp полиморфизм (структурный) [14].
Показаны достоверно более высокое содержание аллеля 4a и частота генотипа 4a/4a у японцев, страдающих артериальной гипертензией и перенесших инфаркт миокарда [15]. Так как в доступной нам литературе мы не нашли работ по изучению данного полиморфизма с СССУ, представляется интересным изучение распространенности аллелей гена eNOS у больных с СССУ, их здоровых родственников по сравнению с контрольной группой.
Материал и методы
Из базы данных кафедры терапии № 1 Красноярского государственного медицинского университета им. проф. В.Ф. Войно-Ясенецкого были отобраны 29 семей с первичным, наследственным СССУ. Среди 29 пробандов было 19 женщин и 10 мужчин, средний возраст составил 52,3±1,6 года (от 26 до 61 года), а также 113 их родственников I, II и III степени родства (68 женщин и 45 мужчин), средний возраст осмотренных родственников 37,9±1,5 года (от 10 до 83 лет). Среди родственников 1-й подгруппы СССУ был выявлен у 55 человек (37 женщин и 18 мужчин), средний возраст 45,38±1,88 года (от 10 до 73 лет).
Электрокардиографические проявления СССУ (СБ, САБ, «синус-арест» с выскальзывающими сокращениями из нижележащих центров автоматизма, пароксизмы фибрилляции предсердий) у пробандов наблюдаются достоверно чаще, чем у их родственников I, II и III степени родства.
Сопутствующие нарушения проводимости сердца у пробандов встречаются достоверно чаще, чем у их родственников: блокада правой ножки пучка Гиса — 13,79 и 2,65% соответственно (р<0,05), атриовентрикулярная блокада I, II, III степени — у 7,14 и 1,92% соответственно (р<0,05), блокада левой ножки пучка Гиса — у 7,14 и 1,92% соответственно (р<0,05), наджелудочковая экстрасистолия — у 24,14 и 12,39% соответственно (р<0,05). Суммарно нарушения ритма и проводимости сердца у пробандов зафиксированы также достоверно чаще (65,52%), чем у родственников этих больных (30,09%).
Частота сопутствующих ССЗ, таких как ишемическая болезнь сердца (ИБС), стенокардия II—III функционального класса, постинфарктный кардиосклероз, сочетание ИБС и гипертонической болезни (ГБ) III стадии достоверно не различалась в группе пробандов СССУ и их родственников. Исключение составили данные по ГБ I—II стадии. Эта патология была выявлена у 5 (35,71%) из 113 родственников и 5 (17,24%) из 29 пробандов (р<0,05). Выявленные сопутствующие ССЗ не могут рассматриваться как причина СССУ, так как электрокардиографические признаки СССУ у этих больных были документированы еще до появления первых признаков ИБС и ГБ.
Всем пробандам и их родственникам I, II, III степени родства было проведено клинико-инструментальное исследование: клинический осмотр, электрокардиография, велоэргометрия, холтеровское мониторирование электрокардиограммы, атропиновая проба, электрофизиологическое исследование (чреспищеводная стимуляция левого предсердия до и после медикаментозной вегетативной блокады), эхокардиоскопия, кардиоритмография.
Молекулярно-генетическое исследование у больных с СССУ и их родственников I, II и III степени родства проводилось в лаборатории молекулярно-генетических исследований терапевтических заболеваний НИИ терапии СО РАМН (Новосибирск).
Для определения полиморфизма гена ADRA2B были взяты образцы крови 213 человек, из которых 75 — больные с диагнозом СССУ, 49 — их здоровые родственники I, II, III степени родства, и 89 человек контрольной группы. Для определения полиморфизма гена eNOS были взяты образцы крови 239 человек, из которых 68 — больные с диагнозом СССУ, 41 — их здоровые родственники I, II, III степени родства и 130 человек контрольной группы.
Контроль подбирали по полу и возрасту из популяционной выборки жителей Новосибирска, обследованных в рамках международного проекта ВОЗ MONICA (Мониторинг заболеваемости и смертности от ССЗ) [16], средний возраст 60±0,1 года, без ССЗ. Исключение диагноза СССУ и других ССЗ состояло в следующем. Основные скрининговые обследования по проекту MONICA проводились стандартными эпидемиологическими и дополнительными методами выявления ССЗ и факторов риска: измерение артериального давления, антропометрия (рост, вес), социально-демографические характеристики, опрос о курении, потреблении алкоголя (частота и типичная доза), уровне физической активности, оценка липидного состава крови (общий холестерин — ОХС; триглицериды — ТГ; и холестерин липопротеидов высокой плотности — ХС ЛВП), опрос для выявления стенокардии напряжения (Rose), электрокардиограмма покоя в 12 отведениях с оценкой по Миннесотскому коду, атропиновый тест для исключения СССУ.
Пробанды с СССУ и их больные родственники были объединены в одну группу, так как исследовалась выборка семей с наследственно обусловленным СССУ. Нам представляется это целесообразным, так как у этих родственников был подтвержден диагноз первичного, наследственно обусловленного СССУ, т.е. все признаки данного синдрома проявлялись у них до возникновения вторичного ССЗ (ИБС, ГБ), которое могло обусловить возникновение данного синдрома. Выборка этих больных наблюдается с 1990 г., когда на ретроспективных электрокардиограммах были выявлены признаки заболевания. ДНК из крови выделяли методом фенолхлороформной экстракции [17, 18].
Генотипирование I/D-полиморфизма гена ADRA2B проводили с помощью полимеразной цепной реакции (ПЦР). Структура праймеров: прямой — 5’-AGGGTGTTTG-TGGGG-CATCT-CC-3’, обратный — 5’-CAAGC-TGAGG-CCGGA-GACAC-TG-3’. Смесь для ПЦР объемом 12,5 мкл включала Трис-HCl (pH 9,0) 75 мM, (NH4)2SO4 20 мM, Тween-20 0,01%, каждого праймера по 0,4 мкM, по 0,24 мМ раствора каждого из четырех dNTP, MgCl2 2,5 мM, 0,6 ед. Тag-полимеразы; 0,5 мкг ДНК. Амплификацию проводили в следующем температурном режиме: 95 °С/1 мин, 68 °С/1 мин, 72 °С/1 мин — 10 циклов, 95 °С/30 с, 68 °С/30 с, 72 °С/30 с — 20 циклов. Наличие продукта ПЦР идентифицировали методом гель-электрофореза в 4% полиакриламидном геле с последующей окраской бромистым этидием. Длина продукта составляла 112 п.н. для инсерционного аллеля и 103 п.н. для делеционного. Таким образом, при генотипе II детектировался только продукт размером 112 п.н., при генотипе DD — размером 103 п.н., при генотипе ID — оба указанных продукта.
Генотипирование I/D-полиморфизма гена eNOS проводили с помощью ПЦР. Структура праймеров: прямой — 5’- AGGCCCTATGGTAGTGCCTT-3’; обратный — 5’-TCTCTTAGTGCTGTGGTCAC-3’. Смесь для ПЦР объемом 12,5 мкл включала: Трис-HCl (pH 9,0) 75 мM, (NH4)2SO4 20 мM, Тween-20 0,01%, каждого праймера по 0,5 мкM, по 0,5 dNTP, MgCl2 1,25 мM, 0,6 ед. Тag-полимеразы, 0,5 мкг ДНК. Амплификацию проводили в следующем температурном режиме: 95 °С/20 с, 65 °С/20 с, 72 °С/20 с — 30 циклов. Наличие продукта ПЦР идентифицировали методом гель-электрофореза в 4% полиакриламидном геле с последующей окраской бромистым этидием. Длина продукта составляла 204 п.н. для нормального аллеля и 177 п.н. для мутантного. Таким образом, при генотипе II детектировался только продукт размером 204 п.н., при генотипе DD — размером 177 п.н., при генотипе ID — оба указанных продукта.
Статистическую обработку данных проводили с использованием пакета программ Statistica 7.0. Первым этапом определяли частоты аллелей и генотипов изучаемых генов-кандидатов. Соответствие распределения аллелей и генотипов равновесию Харди—Вайнберга, сравнительный анализ частот генотипов вышеперечисленных генов с контрольной группой выполняли с использованием критерия χ², двустороннего критерия Фишера.
Результаты и обсуждение
По полиморфизму I/D гена ADRA2B были прогенотипированы 75 больных с СССУ, 49 их здоровых родственников I, II и III степени родства и 89 лиц контрольной группы.
Установлено (рис. 1) достоверное преобладание гомозиготного генотипа по более редкому аллелю DD у больных с СССУ (28%) по сравнению c лицами контрольной группы (8,99%). Однако делеционный вариант аллеля часто встречается у славян (31%) и связан in vivo со снижением поточно-опосредованной дилатации брахиальной артерии и снижением кровотока по коронарным сосудам [19]. У гомозигот DD повышен риск нарушения функции эндотелия, чем объясняется повышение риска развития инфаркта миокарда. Хотя остается неясным вопрос, как влияет I/D-полиморфизм на функцию эндотелия: прямо или опосредованно (через нарушение симпатической активации). Таким образом, изученный генетический маркер может быть использован для выявления предрасположенности к наследственному СССУ.

По полиморфизму 4a/4b гена eNOS были прогенотипированы 68 больных с СССУ, 41 их здоровый родственник I, II и III степени родства и 130 лиц контрольной группы.
Установлено (рис. 2) достоверное преобладание гетерозиготного генотипа 4a/4b у больных с СССУ (41,18%) по сравнению c лицами контрольной группы (25,39%).

В то же время группой украинских ученых были обследованы дети с брадиаритмиями. При этом выявлены достоверные различия по полиморфизму Т786С промотора гена eNOS. Частота патологического аллеля С среди детей встречалась у 34,5% гетерозигот и 15,8% гомозигот по сравнению с 45,8 и 7,14% в контрольной группе. При этом количество патологических гомозигот в группе детей с аритмиями в 2,2 раза превышало таковое в контрольной группе [20]. Однако в европейской популяции аллель 4b гена eNOS встречается значительно чаще, чем аллель с 4 повторами. Распределение частот аллелей в популяции составляет соответственно 4b/4b — 0,41, 4b/4a — 0,46 и 4а/4a — 0,13. У австралийских представителей европеоидной расы и японцев, гомозиготных по аллелю eNOS4а, уровень нитратов и нитритов в крови, напрямую связанный со скоростью выработки NO эндотелием сосудов, достоверно выше, чем у лиц с генотипом 4b/4b. Это свидетельствует о потенциальной роли генотипа 4a/4a как фактора риска развития заболеваний, сопровождающихся нарушением нормальной выработки окиси азота [21].
Мы предполагаем, что полиморфизм промотора гена влияет на транскрипцию мРНК, в то время как полиморфизм экзона определяет белковую структуру и активность фермента. В настоящее время остается неясной роль полиморфизма интрона в регуляции синтеза различных ферментов, так как эта часть гена не кодирует белок (вырезается при формировании спелой РНК). Ввиду того что анализируемый однонуклеотидный полиморфный маркер расположен в некодирующей части гена, наблюдаемая ассоциация может являться результатом как собственного влияния данного полиморфизма на экспрессию гена, так и его сцепления с влияющими на этот процесс аллелями других вариабельных участков гена.
Наше исследование поможет проводить профилактику заболевания на популяционном и индивидуально-семейном уровне.
В условиях кардиологического диспансера, в поликлиниках, медико-генетических консультациях для практической оценки генетического риска развития заболевания (в частности СССУ) возможно использование формулы для определения генетического риска:
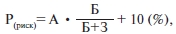
где А — возраст, Б — число больных родственников, З — число здоровых родственников.
Допустим, что у пациента в возрасте 30 лет собраны сведения о 10 кровных родственниках и среди них отмечено 5 случаев ССЗ. Тогда, согласно формуле, априорный риск равен 25%:

Аналогично можно проанализировать риск развития заболевания у этого пациента через 20 лет (апостериорный риск). Тогда, согласно формуле, он равен 35%:
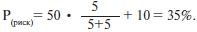
Определение генетического риска для организации здравоохранения означает использование основного принципа семейной медицины, который заключается в том, что основной единицей наблюдения врача является семья. Этот принцип сопоставим с участковым принципом организации поликлинической службы. В практической работе врача возможны следующие виды сбора семейного анамнеза:
• заполнение стандартизированной семейной анкеты;
• заполнение упрощенной родословной.
Применительно к популяции сущность медико-генетического прогноза заключается в выявлении индивидуумов с высоким риском развития заболевания, их дальнейшей диспансеризации и первичной профилактики. Определение генетического риска возникновения какой-либо патологии для организации здравоохранения означает использование основного принципа семейной медицины, который заключается в том, что основной единицей наблюдения врача является семья.
По своей направленности медико-генетический прогноз в популяции взаимосвязан с семейным прогнозом, однако полностью не исчерпывается им, так как в популяции всегда существуют индивидуумы с очень высоким риском развития заболевания и их нельзя выявить посредством больного пробанда при фенотипически здоровых родителях. Генетическое обследование семьи позволяет выявлять патологию СУ на более раннем этапе развития данного синдрома.
Таким образом, проведенное исследование свидетельствует о перспективности дальнейшего поиска генетических факторов, определяющих индивидуальные особенности возникновения СССУ и, в частности, изучения его связи с полиморфизмами генов ADRA2B и eNOS.


