
Синдром Бланда—Уайта—Гарланда (СБУГ) — это врожденный порок сердца, при котором отмечается аномальное отхождение левой коронарной артерии (ЛКА) от ствола легочной артерии (ЛА) (рис. 1).

Первое наблюдение этой врожденной аномалии принадлежит H. Brooks и датируется 1886 г. Впервые подробно описали отхождение ЛКА от легочного ствола М. Abott (1908) и А.И. Абрикосов (1911). Позднее, в 1933 г., американские кардиологи E. Bland, P. White и J. Garland подробно сформулировали клинические и электрокардиографические признаки данной аномалии у грудного ребенка и описали их как синдром, который впоследствии и был назван их именами (BWG-syndrome).
СБУГ является относительно редкой врожденной патологией. Порок выявляют примерно у 1 из 300000 живых новорожденных, что составляет 0,24—0,46% всех врожденных аномалий сердца [1]. Однако некоторые авторы [2] полагают, что эта аномалия коронарных артерий (КА) наблюдается на порядок чаще, чем диагностируется, и является одной из самых частых причин развития инфаркта миокарда (ИМ), сердечной недостаточности (СН) и внезапной смерти у детей первого года жизни. В настоящее время нет эпидемиологических данных о частоте впервые выявленного СБУГ у взрослых пациентов. В мировой литературе описано незначительное количество случаев наблюдения пациентов с СБУГ в зрелом возрасте [3—6].
Эмбриогенез аномального отхождения ЛКА от ЛА до настоящего времени остается неясен и является предметом дискуссии.
В настоящей статье рассмотрен клинический случай впервые выявленного СБУГ у пациентки во взрослом возрасте (25 лет), в том числе обсуждаются особенности консервативного лечения с положительной динамикой состояния на фоне назначенной терапии, а также схема радикального лечения — хирургическая коррекция порока после адекватной подготовки к оперативному вмешательству на фоне сопутствующей патологии.
Анатомические и патофизиологические изменения при СБУГ
Различают 4 варианта этой аномалии: отхождение левой, правой, обеих или добавочной КА от ЛА. Наибольший интерес представляет аномальное отхождение ЛКА, так как аномалии правой и добавочной КА, как правило, не сопровождаются выраженными клиническими проявлениями, а дети с аномальным отхождением обеих КА нежизнеспособны.
Синдром проходит в своем развитии 3 основные патофизиологические фазы [7]: 1-я фаза характеризуется адекватным кровенаполнением ЛКА за счет высокого давления в легочном стволе; 2-я фаза — критическая, обусловлена падением давления в легочном стволе и развитием сети анастомозов между правой коронарной артерией (ПКА) и ветвями ЛКА; 3-я фаза соответствует стадии сформированных анастомозов. На этом, последнем, этапе создается единая сеть коронарного кровообращения с различными уровнями давления. Так, в ПКА систолическое давление равно давлению в аорте и в несколько раз превышает систолическое давление в ЛКА. Поступающая через анастомозы кровь из ПКА в ретроградном направлении идет в ЛКА и затем в ЛА. Таким образом, СБУГ приобретает черты врожденного артериовенозного свища со сбросом крови слева направо. Клинические проявления связаны с фазой снижения давления в ЛА, причем их выраженность зависит от адекватности развивающихся анастомозов, типа кровоснабжения сердца и темпа снижения давления. После рождения у ребенка с СБУГ развивается хроническая гипоксия миокарда, связанная с поступлением венозной, мало насыщенной кислородом крови. Дальнейшее развитие заболевания включает формирование очагов некроза, дистрофии миокарда, снижение сократительной способности сердца, его дилатацию и развитие систолической СН.
Степень нарушения гемодинамики зависит от развитости коллатералей между системой ЛКА и ПКА. Выделяют инфантильный тип порока — с плохо развитыми коллатералями, и взрослый тип — с хорошо развитыми коллатералями. В связи с этим различна и клиническая картина данного порока, которая, прежде всего, обусловлена недостаточностью коронарного кровоснабжения. При инфантильном типе порока больные умирают от обширного ИМ левого желудочка (ЛЖ) в грудном возрасте.
В случае развитых коллатералей постоянная ишемия миокарда быстро приводит к субэндокардиальным и трансмуральным ИМ в области передней стенки и верхушки ЛЖ, нередко в этих зонах могут образоваться аневризмы. Довольно часто формируется недостаточность митрального клапана (МК) вследствие фиброза свободного края створки или деформации передней папиллярной мышцы [8].
Клиническая картина и прогноз
Клинические проявления синдрома обусловлены ишемией миокарда и у большинства детей манифестируют в первые 3 мес жизни. У младенцев отмечаются вялость, бледность кожных покровов, повышенная потливость, рвота, срыгивание, одышка, тахикардия. У отдельных больных первыми признаками болезни являются приступы внезапного беспокойства с появлением тахипноэ, бледности, потливости. Такие приступы возникают обычно во время и после кормления и продолжаются в течение нескольких минут (стенокардия кормления).
При неблагоприятном течении приступы учащаются и в результате тяжелой ишемии миокарда (или ИМ) развивается кардиогенный шок с летальным исходом (85—90% случаев) на первом году жизни ребенка (инфантильный тип). Если ребенок переживает этот критический возраст, то может наступить улучшение с исчезновением ангинозных приступов. Вместе с тем иногда у детей в возрасте 1—2 лет развиваются последствия множественных ИМ с плохо развитыми коллатералями и резко выраженной недостаточностью кровообращения. ЛЖ у таких пациентов практически не выполняет насосную функцию вследствие выраженного кардиосклероза. Это далеко зашедшая стадия необратимых изменений в миокарде, вызывающих инвалидизацию больного. Прогноз в таких случаях неблагоприятный. Нередко возможна смерть в более позднем возрасте на фоне физической нагрузки, часто без предшествующих признаков СН (взрослый тип).
При обследовании ребенка на электрокардиограмме (ЭКГ) можно выявить характерные признаки ишемии (острого ИМ) переднебоковой стенки ЛЖ при нормальном положении или отклонении электрической оси сердца влево. Отмечаются глубокие и уширенные зубцы Q и отрицательные зубцы Т в отведениях I, aVL и левых грудных V5—V6. Кроме того, имеются признаки левожелудочковой гипертрофии (глубокие зубцы S в правых грудных и реже высокие зубцы R в левых грудных отведениях). Сегмент ST обычно ниже изоэлектрической линии.
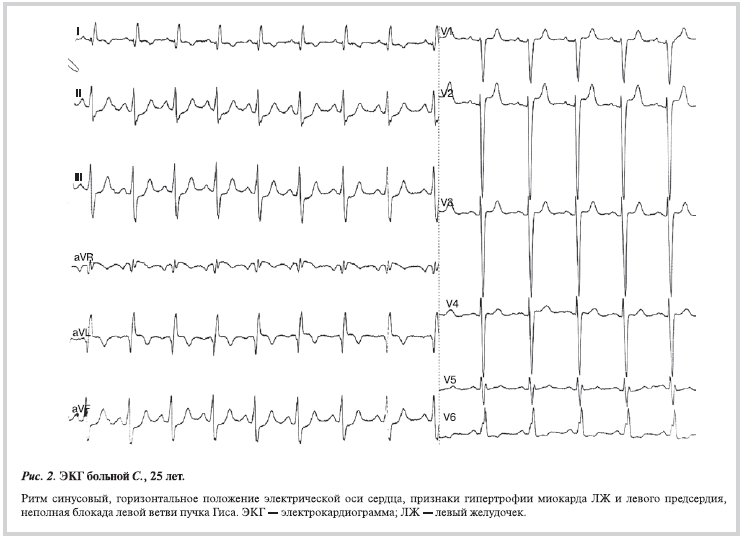
При взрослом типе порока на ЭКГ признаки ИМ могут отсутствовать [9] (рис. 2).
Иногда отмечают более глубокие зубцы Q в I и aVL отведениях, высокие зубцы R в левых грудных отведениях (V4—V6), признаки умеренной гипертрофии миокарда ЛЖ. У ряда больных отмечаются признаки рубцовых изменений в передневерхушечной зоне ЛЖ.
Рентгенологическое исследование не имеет высокого приоритета в диагностическом поиске. Нередко обнаруживается увеличение размеров сердца, чаще его левых отделов.
Эхокардиография (ЭхоКГ) может помочь в диагностике данного порока сердца. Задачей этого исследования является визуализация корня аорты с устьями КА. Увидеть изображение ЛКА у легочного ствола труднее, но возможно при правильном положении датчика.
Мультиспиральная компьютерная томография (МСКТ) и коронарография (КГ) с вентрикулографией являются наиболее чувствительными и информативными методами диагностики аномального отхождения КА. Они позволяет четко определить местоположение устьев КА, а также выявить сопутствующую патологию сердца и сосудов [10]. Селективная КГ визуализирует единственную расширенную ПКА и быстрое ретроградное контрастирование системы ЛКА со сбросом крови в ЛА и выявляет тип коронарного кровообращения (левый или правый). Это последнее обстоятельство, по данным ряда авторов [7], имеет значение для прогноза заболевания (более благоприятный при правом типе).
Описание клинического случая
В феврале 2011 г. в кардиологическое отделение клинической больницы № 83 ФМБА России была госпитализирована пациентка С., 25 лет. При поступлении основными жалобами пациентки были быстрая утомляемость (выраженная усталость при подъеме на 1 этаж); общая слабость; тахикардия в покое и при нагрузке. Кроме того, пациентку беспокоили дискомфорт и ощущение тяжести в области сердца; снижение массы тела на 4—5 кг в течение года, повышенная тревожность и нарушение сна.
При опросе удалось выяснить, что, начиная с недельного возраста, больная находилась под наблюдением у педиатра в связи с предполагаемой патологией сердца.
В грудном возрасте у пациентки был диагностирован фиброэластоз эндокарда, а впоследствии по данным ЭКГ и клинической картины — врожденный кардит.
Следует отметить, что больная С. родилась от первой беременности, протекавшей с токсикозом в I триместре, мать пациентки в I триместре перенесла ОРВИ. Девочка родилась с массой 2700 г, рост — 49 см, с оценкой по шкале Апгар 8 баллов, прививка БЦЖ выполнена в роддоме. Период новорожденности протекал без особенностей.
В дошкольном и раннем школьном возрасте ведущим диагнозом пациентки была дилатационная кардиомиопатия с увеличением размеров левых отделов сердца и пролапсом МК с регургитацией III—IV степени. В 14-летнем возрасте девочка находилась под наблюдением кардиолога с основным диагнозом «постмиокардический кардиосклероз». За год до настоящей госпитализации пациентка повторно обратилась за медицинской помощью в связи с развитием трех эпизодов синкопального состояния. По данным холтеровского мониторирования ЭКГ зафиксирована частая желудочковая экстрасистолия, в связи с чем был назначен амиодарон по стандартной схеме.
При поступлении объективный статус больной С. без особенностей. При физикальном обследовании было выявлено увеличение пульса в покое до 90 в минуту и систолический шум на верхушке сердца при аускультации.
По данным лабораторных методов исследования общий анализ крови, биохимические показатели крови, кардиоспецифические ферменты в рамках физиологической нормы. Мы обратили внимание на повышение уровня мозгового натрийуретического пептида до 565 пг/мл, а также признаки тиреотоксикоза с повышением уровня свободного трийодтиронина до 8,33 пг/мл и снижением уровня тиреотропного гормона (ТТГ) до 0,004 мкМЕд/мл,свободного тироксина— до 1,91 пмоль/л.
Данные инструментальных исследований не позволили предположить наличие у пациентки врожденного порока сердца. На ЭКГ отмечалась синусовая тахикардия, признаки гипертрофии ЛЖ и левого предсердия с нарушением кровоснабжения переднеперегородочной области ЛЖ. Результаты холтеровского мониторирования ЭКГ были без особенностей, зарегистрировано 8 желудочковых экстрасистол.
Однако при ЭхоКГ отмечались диастолическая дисфункция миокарда по рестриктивному типу и гипокинез средне-переднеперегородочного и заднеперегородочного сегментов, некоторая дискинезия верхушки, фракция выброса 37—40%. При цветовом допплеровском картировании определялись узкие линейные потоки в миокарде желудочков шириной до 0,8 см (предположительно расширенные сосуды сердца). ЛЖ 5,7 см, левое предсердие — 91 мл, эксцентрическая гипертрофия ЛЖ, масса миокарда 273 г. Отмечались также пролабирование обеих створок МК, миксоматозная дегенерация створок МК, митральная регургитация IV степени.
На основании структурных изменений, обнаруженных по данным ЭхоКГ, больной была рекомендована КГ, результаты которой помогли установить диагноз.
КГ позволила выявить аномальное отхождение ствола ЛКА от корня ЛА с формированием коронарного синдрома обкрадывания (рис. 3).

ПКА отходит типично, просвет ее расширен и через мощные септальные и апикальные перетоки отмечается поступление рентгенконтрастного вещества в неизмененные ветви и ствол ЛКА, со сбросом контрастного вещества в ствол ЛА.
Таким образом, на основании клинической картины заболевания и данных лабораторно-инструментальных исследований пациентке установлен окончательный клинический диагноз: врожденный порок сердца: синдром Бланда—Уайта—Гарланда. Осложнения: ХСН I степени, II функционального класса по классификации NYHA. Митральная недостаточность III—IV степени. Сопутствующие заболевания: кордарон-индуцированный тиреотоксикоз, средней степени тяжести, медикаментозная декомпенсация. Тиреотоксическое сердце.
В связи с развитием кордарон-индуцированного тиреотоксикоза больной была рекомендована следующая терапия: тиамазол 40 мг/сут, ивабрадин 15 мг/сут, метапролола сукцинат 25 мг/сут. Кроме того, проведено 5 сеансов лечебного плазмафереза.
На фоне подобранной терапии отмечались улучшение общего состояния, нормализация пульса, исчезновение ангинозных приступов и повышение толерантности к физической нагрузке. По данным дополнительного обследования выявлена положительная динамика результатов ЭхоКГ (увеличение фракции выброса до 52%, уменьшение митральной регургитации с IV до III степени), а также и положительная динамика уровня гормонов щитовидной железы и ТТГ (снижение свободного трийодтиронина до 4,44 пг/мл и повышение свободного тироксина до 2,87 пмоль/л на момент выписки из стационара).
Обсуждение
Вопросы диагностики и лечения врожденных пороков сердца с транспозицией артерий продолжают оставаться актуальными в связи с трудностями диагностики как в ранние периоды жизни пациента, так и во взрослом возрасте. К сожалению, в весьма большом проценте случаев СБУГ может не проявляться клинически и дебютировать ИМ или внезапной сердечной смертью на фоне развития угрожающих жизни желудочковых аритмий и выявляется лишь на этапе патологоанатомического вскрытия. Отмечается также крайне низкая распространенность данной аномалии у взрослых, так как большинство детей с СБУГ умирают в раннем возрасте. Кроме того, СБУГ имеет «маски» других заболеваний, таких как миокардит, постмиокардитический кардиосклероз, дилатационная кардиомиопатия, пролапс и/или недостаточность МК, ИМ (что, в частности, подтверждается анамнезом пациентки С.).
В связи с изложенным особо важное значение имеют методы диагностики, которые позволяют точно верифицировать врожденный порок сердца: КГ и МСКТ. Именно КГ позволила диагностировать СБУГ у пациентки С. По данным КГ у больной С. был выявлен правый тип кровоснабжения миокарда с хорошо развитыми мощными коллатералями ПКА. Очевидно, эти анатомические особенности коронарного кровообращения позволили компенсировать нарушения гемодинамики, обусловленные атипичным отхождением ЛКА от ствола ЛА, в связи с чем у больной в возрасте 25 лет отсутствовали выраженные признаки СН.
В случае успешной диагностики СБУГ пациенты должны быть незамедлительно направлены в кардиохирургический стационар для выбора тактики хирургического лечения и проведения операции в минимально возможные сроки. Выбор оптимальной тактики лечения имеет определяющее значение для отдаленного прогноза у этой категории пациентов.
Не вызывает никаких сомнений, что наличие у пациента СБУГ служит абсолютным показанием к радикальному методу лечения, и в настоящее время общепринятой является хирургическая коррекция порока сразу после постановки диагноза. Существует несколько вариантов хирургического лечения, но задача этих методов одна — создание адекватной перфузии (кровоснабжения) миокарда в бассейне ЛКА. Возможны следующие варианты хирургических вмешательств [2]:
1) перевязка устья аномальной КА для ликвидации steal-синдрома (синдрома обкрадывания). Операцию проводят только в случае хорошо развитых коллатералей. Вся нагрузка ложится на нормальную ПКА;
2) аортокоронарное шунтирование аномально отходящей ЛКА;
3) транслокация устья ЛКА в аорту. Целью этой операции является перенос устья ЛКА на единой площадке в аорту;
4) создание внутрилегочного тоннеля (операция Takeuchi);
5) маммарокоронарный анастомоз с аномально отходящей ЛКА.
В случае развития митральной недостаточности (из-за недостаточного кровоснабжения папиллярных мышц МК) также проводят коррекцию клапанного порока. Согласно данным литературы [11, 12], важную роль в возникновении недостаточности МК играют 2 фактора: ишемический и эмбриологический. В основе эмбриологического компонента лежат нарушения закладки отделов сердца, которые заключаются в развитии «доминантности» ЛЖ вследствие смещения задней межжелудочковой перегородки в полость ПЖ, что приводит к увеличению фиброзного кольца левого атриовентрикулярного клапана [13]. Дисфункция МК является неотъемлемой частью СБУГ и в отсутствие своевременной хирургической коррекции приводит к развитию ХСН, что ухудшает прогноз и качество жизни пациентов. У детей первого года жизни, включая новорожденных, операцией выбора является аннулопластика по Reed или ее модификации. У детей старшего возраста используют шовную пластику МК с резекцией отдельных хорд. При необходимости в многокомпонентной реконструкции операцией выбора считается замена клапана искусственным протезом [13].
Первая успешная операция по поводу СБУГ была проведена 3-месячной девочке в 1968 г. B.W. Meyer и заключалась в наложении подключично-коронарного анастомоза [14]. В 1974 г. W.H. Neches и соавт. впервые осуществили прямую имплантацию аномально отходящей ЛКА в аорту [15].
По данным литературы, наиболее часто применяемым методом хирургического лечения является прямая реимплантация устья ЛКА в аорту [16]. Именно этот вариант хирургической коррекции СБУГ был выполнен пациентке С. в сочетании с протезированием МК механическим протезом. Несколько менее эффективны шунтирование ЛКА с помощью внутригрудных артерий или соединение аорты и ЛКА с помощью туннеля внутри легочного ствола (операция Takeuchi) [17, 18].
Заключение
Синдром Бланда—Уайта—Гарланда — редкое заболевание, относящееся к категории врожденных пороков сердца и характеризующееся аномальным отхождением левой коронарной артерии от легочного ствола. Трудности диагностики синдрома Бланда—Уайта—Гарланда у взрослых пациентов обусловлены отсутствием специфических признаков заболевания по данным электрокардиографии, эхокардиографии, сцинтиграфии миокарда. «Золотым стандартом» диагностики являются коронарография и мультиспиральная компьютерная томография, которые проводятся молодым пациентам довольно редко ввиду отсутствия четких показаний, возможности выполнения данных методик по причине недоступности во многих стационарах, а также дороговизны исследования.
У пациентки С., 25 лет, диагноз синдрома Бланда—Уайта—Гарланда был верифицирован с помощью коронарографии, что позволило оказать своевременную медицинскую помощь и провести хирургическую коррекцию врожденного порока сердца.



{kind=link}