
Атеросклеротические повреждения возникают вследствие отложения в сосудистой стенке липидов,
преимущественно холестерина (ХС), главным источником которого служат липопротеиды низкой плотности (ЛНП). Высокое содержание ХС в плазме крови является фактором риска возникновения и развития атеросклероза, однако высокий уровень ХС в ЛНП не всегда приводит к возникновению сердечно-сосудистых заболеваний, что указывает на важную роль других нарушений метаболизма ЛНП в атерогенезе. Считается, что атеросклеротические повреждения сосудов происходят в результате неконтролируемого поглощения липидов моноцитами-макрофагами стенки сосудов, при этом перегруженные липидами клетки погибают путем апоптоза, формируя липидное ядро атероскле-
ротической бляшки.
В норме ЛНП поглощаются клетками стенки сосуда с участием рецепторов ЛНП, концентрация которых на мембране зависит от внутриклеточного содержания ХС [1]. Таким образом, поглощение нативных ЛНП контролируется по принципу отрицательной обратной связи, однако проникновение в клетки ЛНП с измененной структурой может происходить по другим механизмам, не зависящим от уровня ХС в клетке.
При атеросклерозе в условиях усиленного накопления активных форм кислорода и их метаболитов
одновременно развивается недостаточность систем антиоксидантной защиты организма, что приводит к возникновению окислительного стресса [2]. Это создает условия для окисления липидов частиц ЛНП, причем окислительная деструкция образующихся липопероксидов ведет к накоплению вторичных карбонильных продуктов. Окисление полиненасыщенных жирнокислотных остатков фосфолипидов наружного слоя ЛНП сопровождается изменением заряда частиц, что может вызывать их агрегацию [3]. Модификация белковой составляющей ЛНП — апопротеина (апо) В-100 может происходить вследствие реакции свободных аминогрупп лизиновых и аргининовых остатков с альдегидными группами природных дикарбонилов, что сопровождается образованием внутри- и межмолекулярных
сшивок типа шиффовых оснований [4] и также может вызвать формирование конгломератов частиц ЛНП [3]. Агрегаты частиц ЛНП захватываются клетками стенки сосуда преимущественно путем фагоцитоза [5, 6]. Накопление липидов в макрофагах тесно связано со степенью агрегации ЛНП и поглощение агрегированных ЛНП может быть более эффективным. Так, подвергшиеся окислительной модификации, но неагрегированные частицы ЛНП, захватываются макрофагами в значительно меньшей степени, чем агрегаты модифицированных частиц [5]. В то же время агрегация
немодифицированных ЛНП, вызванная их интенсивным перемешиванием, может индуцировать накопление липидов в культивируемых макрофагах [5, 7].
Важную роль в слипании частиц играет модификация липидной части наружного слоя ЛНП, хотя модификация белковых компонентов ЛНП также может иметь значение при образовании агрегатов [3]. Химическая модификация белковой части ЛНП (апоВ-100) может приводить к поглощению ЛНП макрофагами по другому механизму — через скэвенджер-рецепторы, активно захватывающие поврежденные белковые молекулы [8—10]. При взаимодействии апоВ-100 с карбонильными соединениями белок теряет положительные заряды, изменяет свою конформацию и перестает опознаваться ЛНП-рецепторами [8]. В отличие от ЛНП-рецепторов, захват ЛНП через скэвенджер-рецепторы не регулируется по принципу отрицательной обратной связи, в связи с чем модифицированные ЛНП захватываются макрофагами более активно [11].
При атеросклерозе in vivo наиболее вероятна модификация частиц ЛНП в результате их взаимодействия с конечными продуктами перекисного окисления липидов, такими как малоновый диальдегид (МДА) [4]. Гипергликемия при сахарном диабете (СД) может вызывать изменения в окислительном метаболизме глюкозы и развитие карбонильного стресса, т.е. интенсивное накопление низкомолекулярных дикарбонилов, близких по структуре к МДА — метилглиоксаля и глиоксаля [12, 13].
СД — фактор риска развития атеросклероза, причем при сопутствующем СД отмечено резкое прогрессирование атеросклеротических повреждений сосудов [14]. Кроме того, показано, что уровень липогидропероксидов в ЛНП плазмы крови у больных с СД в 3 раза выше, чем у пациентов с ишемической болезнью сердца [2]. Таким образом, гипергликемия вызывает развитие не только карбонильного (накопление низкомолекулярных альдегидов), но и выраженного окислительного стресса (накопление липопероксидов), что может провоцировать резкое увеличение атерогенности ЛНП.
В настоящей работе исследовали влияние карбонильной модификации ЛНП на свойства частиц,
определяющие их повышенную атерогенность, такие как образование межмолекулярных сшивок в апоВ100, окисляемость частиц ЛНП и их способность к последующей агрегации.
Материал и методы
ЛНП выделяли из объединенных образцов плазмы крови 3 здоровых доноров двухстадийным ультрацентрифугированием в градиенте плотности NaBr на ультрацентрифуге Beckman L-8 (США), используя ротор 50Тi [15], после чего ЛНП диализовали против 2000 объемов изотонического фосфатного буфера рН 7,4 при температуре 4 ºС в течение 18 ч. Концентрацию белка в образцах ЛНП определяли по методу Лоури. Модификацию ЛНП проводили в течение 3 ч при температуре 37 ºС в изотоническом фосфатном буфере рН 7,4, содержащем 1 мкмоль МДА, глиоксаля или метил глиоксаля на 100 мкг апоВ-100, а затем от избытка альдегидов освобождали путем диализа как описано выше. МДА получали путем кислотного гидролиза 1,1,4,4-тетраэтоксипропана [4]. Суммарный поверхностный заряд нативных и модифицированных ЛНП оценивали с помощью электрофореза в 1% агарозном геле [16]. Для оценки модификации апоВ100 проводили электрофорез нативных и модифицированных ЛНП в полиакриламидном геле (ПААГ) с 0,1% додецилсульфатом Na (4% концентрирующий, 6% разделяющий гели).
Кинетику свободнорадикального окисления нативных и модифицированных ЛНП (50 мкг белка/мл)
исследовали в изотоническом фосфатном буфере рН 7,4 при температуре 37 ºС в присутствии 30 мкМ
CuSO4 по накоплению первичных продуктов окисления — липогидропероксидов (конъюгированных
диенов), определяя увеличение оптической плотности при 233 нм на спектрофотометре Hitachi 220A
(Япония).
Степень агрегации ЛНП оценивали на двухканальном агрегометре LA220 (НПФ БИОЛА, Россия) [15],
регистрируя флуктуацию светопропускания луча лазерного света с длиной волны 860 нм. Метод основан на том, что относительная дисперсия колебаний оптической плотности, вызванных случайными изменениями в количестве частиц, попадающих в оптический путь лазерного луча, отражает отклонения от их среднего размера, т.е. степень их агрегации. Для исследования агрегационной способности образцы ЛНП (50 мкг белка/мл) инкубировали при температуре 37 ºС в изотоническом фосфатном буфере рН 7,2 либо в той же среде, но в присутствии 30 мкмоль МДА
или метилглиоксаля. Предварительно образцы ЛНП освобождали от имеющихся ассоциатов, пропуская
через фильтр с порами диаметром 0,45 мкм. В ряде опытов в среду инкубации дополнительно вносили
супероксиддисмутазу (СОД) из расчета 50 ед/мл.
Все используемые реагенты были фирмы Sigma (США).
Результаты
Взаимодействие с альдегидами уменьшает суммарный поверхностный заряд частиц, о чем свидетельствуют увеличение электрофоретической подвижности ЛНП в агарозном геле (рис. 1). Взаимодействие с МДА приводит к наибольшему изменению заряда частиц, тогда как инкубация ЛНП с глиоксалем и метилглиоксалем оказывает менее выраженное влияние на поверхностный заряд частиц ЛНП. Кроме того, модификация ЛНП дикарбонильными соединениями приводит к тому, что не все молекулы апоВ100 входят в относительно плотный разделяющий гель, очевидно, вследствие образования внутри- и межмолекулярных сшивок, как видно из результатов электрофореза ЛНП в ПААГ (рис. 2). В результате взаимодействия с исследованными альдегидами лишь около 50% частиц ЛНП, модифицированных глиоксалем и метилглиоксалем, входят в разделяющий гель (42,1±6,1% и 50,3±6,7% соответственно, относительно немодифицированных ЛНП), тогда как после модификации МДА практически все частицы ЛНП остаются на границе концентрирующего и разделяющего гелей (см. рис. 2).

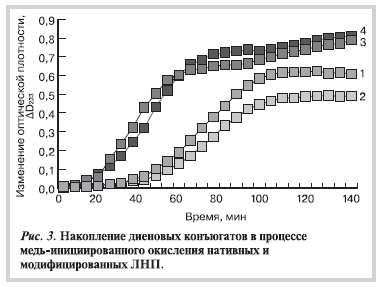
Как видно на рис. 3, после модификации апоВ-100 глиоксалем и метилглиоксалем окисление липидного
компонента ЛНП (накопление липогидропероксидов) идет более интенсивно: уменьшается продолжительность лаг-фазы, возрастают максимальная скорость окисления и суммарная степень окисленности частиц. В то же время модификация апоВ-100 в присутствии МДА, напротив, снижает окисляемость ЛНП (рис. 3).
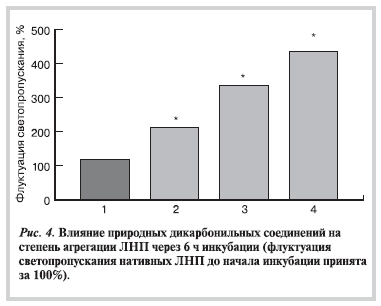
Взаимодействие ЛНП со всеми исследованными дикарбонилами приводит к усилению агрегации частиц
ЛНП, однако размеры агрегатов частиц, образующихся в процессе инкубации, существенно различались в зависимости от использованных альдегидов (рис. 4). Присутствие МДА в среде инкубации ЛНП оказывало наименьшее влияние на скорость агрегации, тогда как присутствие метилглиоксаля в наибольшей степени ускоряло агрегацию.
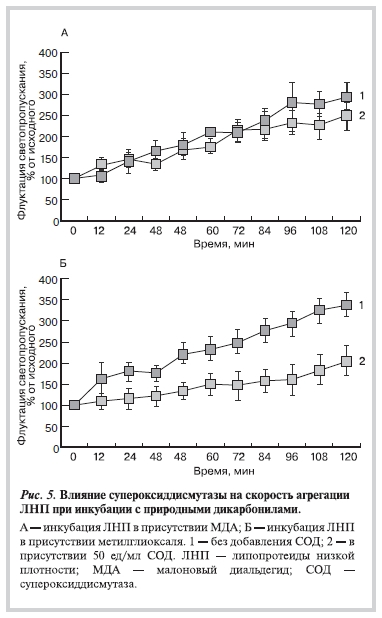
При этом добавление СОД не оказывало достоверного влияния на скорость агрегации ЛНП в присутствии МДА (рис. 5, А), но существенно снижало скорость агрегации ЛНП в присутствии метилглиоксаля (рис. 5, Б).
Обсуждение
Согласно общепринятой теории окислительный стресс инициирует окисление ненасыщенных липидов ЛНП активными формами кислорода, а образующиеся в результате этого липогидропероксиды
подвергаются дальнейшей окислительной деструкции, приводя к накоплению вторичных продуктов
свободнорадикального окисления, преимущественно МДА [4]. Окисление глюкозы, в свою очередь, может способствовать накоплению гомолога МДА глиоксаля и изомера МДА метилглиоксаля [13]. Установлено, что модификация апоВ-100 вторичными продуктами свободнорадикального окисления липидов и глюкозы приводит к увеличению атерогенности частиц и их усиленному захвату клетками стенки сосуда [8, 17], однако вопрос о том, способна ли подобная модификация влиять на дальнейшие окислительные процессы в ЛНП, до настоящего времени не изучен.
Модификация ЛНП дикарбонильными соединениями сопровождается увеличением подвижности частиц в агарозном геле, что свидетельствует об уменьшении суммарного поверхностного заряда частиц,
вероятно, вследствие модификации положительно заряженных аминокислотных остатков апоВ-100 под
действием дикарбонилов (см. рис. 2). Результаты электрофореза в ПААГ также свидетельствуют в пользу того, что взаимодействие ЛНП с дикарбонилами может вызывать значительные изменения белковой компоненты частиц, приводя к образованию внутрии межмолекулярных сшивок, значительно меняющих конформацию апоВ-100 (см. рис. 3). Как следует из полученных нами результатов, модификация ЛНП дикарбонилами оказывает существенное влияние на их дальнейшее окисление (см. рис. 4). МДА, модифицирующий апоВ-100 ЛНП в большей степени, чем глиоксаль и метилглиоксаль (см. рис. 2 и 3), замедляет процессы свободнорадикального окисления липидов наружного слоя ЛНП (см. рис. 4). В то же время глиоксаль и метилглиоксаль, модифицирующие апоВ-100 в меньшей степени, чем МДА (см. рис. 2 и 3), вызывают значительное ускорение свободнорадикальных реакций в ненасыщенных липидах модифицированных альдегидом ЛНП (см. рис. 4). Таким образом, накапливающиеся при СД глиоксаль и метилглиоксаль [13] изменяют структуру белкового компонента ЛНП в меньшей степени, чем МДА. Однако модификация ЛНП этими альдегидами может усиливать дальнейшее окисление ненасыщенных липидов наружного слоя частиц ЛНП, способствуя развитию окислительного стресса. Можно полагать, что увеличение жесткости белковой структуры ЛНП под действием МДА создает большие структурные препятствия дальнейшему окислению частиц, тогда как частичное «сшивание» молекул белка под действием глиоксаля и метилглиоксаля облегчает перекисное окисление ненасыщенных липидов наружного слоя ЛНП.
Добавление метилглиоксаля и глиоксаля в среду инкубации ускоряет образование агрегатов ЛНП в
большей степени, чем добавление МДА (см. рис. 5), что хорошо согласуется с данными по влиянию этих
дикарбонилов на окисляемость частиц ЛНП (см. рис. 4). Действительно, при образовании агрегатов ЛНП важную роль играет структурное изменение наружного липидного слоя частицы, в то время как модификация белковой части имеет меньшее значение [18]. Как показано нами, метилглиоксаль и глиоксаль способствуют окислению липидов наружного слоя частицы, тогда как МДА, напротив, препятствует развитию окислительных процессов в липидном компоненте ЛНП (см. рис. 4). В результате окисления ненасыщенных липидов наружного слоя теряется целостность частицы ЛНП, что и вызывает агрегацию частиц [3, 18].
Результаты данной работы согласуются с опубликованными нами данными о генерировании супероксида при взаимодействии лизиновых остатков с метилглиоксалем [19]. Действительно, вторичное образование супероксидного радикала в процессе альдегидной модификации белка может инициировать продолжение цепного свободнорадикального окисления ненасыщенных липидов ЛНП, приводя к дальнейшему накоплению МДА и других карбонильных соединений. Удаление супероксида из среды инкубации снимает некоторые негативные эффекты, возникающие при модификации апоВ-100 метилглиоксалем: добавление СОД значительно уменьшает агрегацию модифицированных метилглиоксалем ЛНП, при этом скорость агрегации оказывается ниже уровня агрегации под действием МДА (рис. 6).
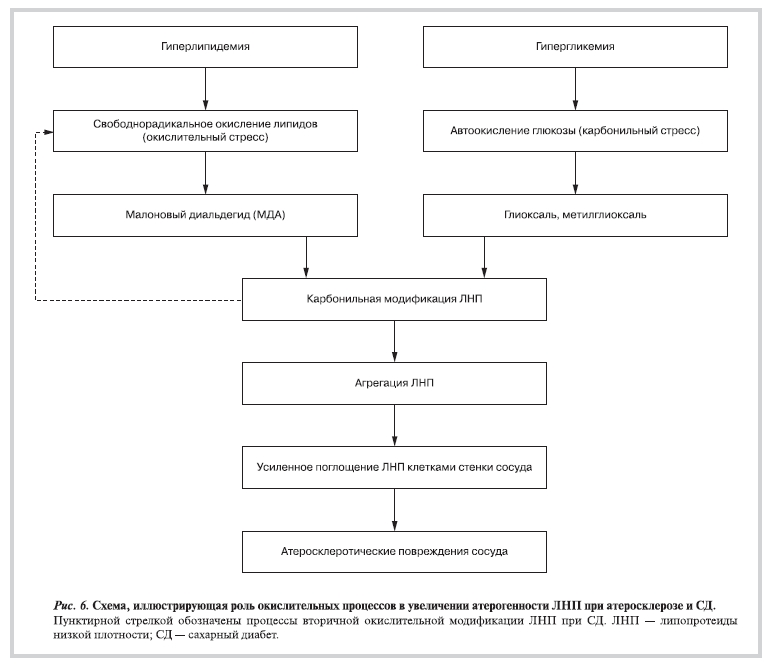
Рисунок 6. Схема, иллюстрирующая роль окислительных процессов в увеличении атерогенности ЛНП при атеросклерозе и СД.
Следовательно, окислительные изменения в наружном липидном слое ЛНП и альдегидзависимая модификация белкового компонента частиц тесно взаимосвязаны. Более того, модификация белка под
действием вторичных продуктов свободнорадикального окисления способна инициировать дальнейшие окислительные превращения частиц ЛНП. Этот процесс может быть особенно значим при СД, так как образующиеся при окислении глюкозы альдегиды (глиоксаль и метилглиоксаль) являются более мощными агентами, вызывающими агрегацию ЛНП. Более того, в процессе взаимодействия с белком таких дикарбонилов, как метилглиоксаль, происходит генерирование активных форм кислорода, способных индуцировать интенсификацию свободнорадикальных реакций окисления ЛНП, приводящих к окислительному стрессу. Последовательность событий, приводящих к атерогенной модификации частиц ЛНП при атеросклерозе и СД, представлена на рис. 6.
Таким образом, наши данные указывают на возможность существования автокаталитического механизма вторичной окислительной модификации ЛНП, увеличивающей их атерогенность, в условиях диабетической гипергликемии. Исходя из этих представлений можно предполагать единый молекулярный механизм атерогенных повреждений стенки сосудов при атеросклерозе и СД, включающем дополнительные автокаталитические реакции модификации ЛНП при СД.



{kind=link}