
Сердечная недостаточность (СН) — сложный клинический синдром, для которого характерны прогрессирующая и практически необратимая потеря сократительной способности миокарда и соответственно снижение снабжения тканей и органов оксигенированной кровью. Чаще всего СН предстает завершающей стадией течения разных по своей этиологии поражений сердца (ишемическая болезнь, атеросклероз, гипертоническая болезнь, недостаточность клапанов, кардиомиопатии, вирусные миокардиты и выявленные в последнее время генные аномалии белков мышечных саркомеров) в пожилом и старческом возрасте. На протяжении более столетия
понимание патогенеза СН усложнялось и достраивалось. Первоначально объяснение развития СН основывалось на кардиоренальной модели, затем возникла гемодинамическая модель, на смену которой пришла нейрогормональная модель патогенеза [1]. Утверждению последней способствовали, прежде всего, экспериментальные данные, показавшие возможность смоделировать СН, изменяя до патофизиологического уровня концентрации соответствующих нейрогормонов. Использование в терапии СН препаратов, противодействующих проявлению активности нейрогормонов, свидетельствует о клиническом улучшении состояния пациентов, и в настоящее время их комбинации активно рекомендуются на основе доказательств многоцентровых исследований как наиболее эффективные для лечения СН.
Однако ясно, что нейрогормональная модель также недостаточна для объяснения патогенеза СН: терапевтический эффект антагонистов нейрогормонов либо их рецепторов непродолжителен, и многие нейрогормоны и цитокины продуцируются самими кардиомиоцитами либо другими клеточными компонентами миокарда, действуя по аутокринному или паракринному способу. Наконец, самое главное — в последнее десятилетие различные направления исследований миокарда, в том числе
данные геномики и протеомики, показали сложность и множество путей и молекулярных механизмов, ассоциирующихся с нарушением сократительной функции миокарда и СН [1]. Эти механизмы изменяют как процессы жизнедеятельности и функционирования клеточных элементов самого миокарда, так и реакцию на них других органов и систем организма, формируя патогенетический континуум, обусловливающий прогрессирование СН. В этом патогенетическом континууме проявление клинических симптомов СН представляется как заключительная и необратимая стадия сформировавшегося патологического процесса, когда резервы компенсаторных механизмов организма оказываются недостаточными и перепрограммирование транскрипции генома, прежде всего в кардиомиоцитах, уже необратимо.
Новое понимание патогенеза СН выдвигает в качестве необходимости выявление более ранних ее признаков, т.е. диагностирование СН до возникновения ее клинических симптомов, и выбор новых системных терапевтических мишеней. Диагностика возникающей СН реальна, по-видимому, лишь на основе биомаркеров нового поколения (геномики и протеомики). Даже выявление начала ухудшения эхокардиографических показателей может оказаться запоздалым для успешного противостояния прогрессированию СН. Как и в онкологии, ранняя диагностика в случае СН представляется важной хотя бы потому, что прогноз СН хуже прогноза большинства раковых заболеваний. Что же касается новых тенденций изучения СН, то их мишенями являются не только участники компенсаторных механизмов, но и зарождающиеся под влиянием повреждающих сигналов процессы транскрипционного перепрограммирования кардиомиоцитов [2], эндогенные механизмы восстановления кардиомиоцитов [3, 4].
Рассмотрим некоторые общие патогенетические аспекты СН, обращая особое внимание на их особенности при старении и на биомаркеры, выявление которых расширяет наше понимание патогенеза СН.
Патогенетический континуум
Неудивительно, что, будучи финальной стадией различных по своей этиологии заболеваний сердца, СН предстает клинически сильно различающимися фенотипическими проявлениями. Однако общими ее признаками служат прогрессирующее снижение сократительной способности миокарда и, соответственно, фракции выброса, расширение полостей желудочков и уменьшение толщины их стенок, что приводит к изменению периферической циркуляции и к дисфункции эндотелия, вызывая нарушение регуляции водно-солевого обмена и функционирования всех систем и органов (сердечно-сосудистой, скелетной мускулатуры, нейроэндокринной, гомеостатической, иммунной, почек). Ограничения в размерах публикации позволяют рассмотреть кратко лишь изменения в самом миокарде. Сложность этих сдвигов частично проиллюстрирована на рис.1, отображающем формирование кардиоренального синдрома при СН [5].
Рисунок. Симптоматическая схема кардиоренального синдрома (по[5]).
Исключая ситуации острого возникновения СН, например, при крупноочаговом инфаркте миокарда (ИМ) либо вирусном миокардите, клиническому проявлению ее патогенетического континуума предшествуют длительное, нередко многолетнее, накопление молекулярных, клеточных и системных патологических изменений, и наиболее подвержены ей люди пожилого и преклонного возраста.
Если раньше объяснение патогенеза СН затрагивало лишь ее поздние проявления — нарушенную функцию левого желудочка (ЛЖ) и форсированную неблагополучную гемодинамику, то ныне акценты сместились к самым ранним истокам СН. Полагают, что миокардиальный стресс (например, сигналы увеличения давления и объемной нагрузки, окислительный стресс, острое повреждение при ИМ) активирует механические и нейрогуморальные сигнальные каскады, которые запускают формирование патогенетического континуума, ведущего к развитию гипертрофии и ремоделированию сердца. Условно в этом континууме выделяют 3 последовательные стадии: адаптивную, компенсаторную и стадию клинических проявлений.
Реагируемыми мишенями перекрывающихся каскадов сигнальной трансдукции при СН являются транскрипционные факторы, коактиваторы и корепрессоры генной экспрессии кардиомиоцитов, эффекторные механизмы (сопряжение возбуждение—сокращение, сократительный аппарат, образование энергии, мобилизация Са2+, метаболизм, рост и апоптоз), которые достигают наибольших изменений при дисфункции желудочков, а также нейрогуморальные реакции, вовлекающие симпатическую и парасимпатическую части вегетативной нервной системы, центральные механизмы регуляции [6]. Наследственные мутации при кардиомиопатиях затра гивают многие белки миокарда и вовлекают сходные каскады событий, следствием которых является миопати-
ческий фенотип [7].
Прямые эффекты стрессовых воздействий на многие, если не все, механизмы трансдукции, которые вовлекаются при СН, конвергируют на клеточном ядре, оказывая долгосрочные влияния практически на каждый аспект функции сердца через усиление экспрессии генов, меняя фенотип кардиомиоцитов. Наиболее ярко это проявляется в повышенном синтезе белков, увеличении структуры саркомеров и измененной транскрипции генов. Большое количество транскрипционных факторов являются медиаторами изменений экспрессии генов при гипертрофическом ответе [2]. Патологические стимулы активируют рост клеток посредством нескольких механизмов, в числе которых: 1) активация клеточных сигнальных систем непосредственно механической нагрузкой; 2) стимуляция механической нагрузкой высвобождения локально действующих секретируемых факторов; 3) ответная активация нейрогуморальных путей на гемодинамический стресс, что обычно ведет к высвобождению пара- и эндокринных факторов [6].
Этот гипертрофический ответ развертывается для противостояния кардиомиоцитов нагрузке и в краткосрочном плане считается адаптивным. Однако со временем молекулярные изменения, которые возникли из-за хронической стимуляции этих сигнальных путей, могут привести к нарушенной функции кардиомиоцитов и к их утрате в результате апоптоза, аутофагии и некроза. Формирующееся нарушение увеличивает нагрузку на оставшиеся кардиомиоциты, поддерживая активацию гипертрофических каскадов, и приводит к порочному циклу потери миоцитов и нарушенной функции миокарда, приводя в итоге к СН [2, 8]. Подтверждением идеи, что гипертрофический ответ является центральным в патогенезе СН, служит, с одной стороны, проявление гипертрофии кардиомиоцитов практически при всех заболеваниях сердца, и, с другой стороны, терапия, уменьшающая ремоделирование ЛЖ и гипертрофию миокарда, эффективно продлевающая продолжительность жизни пациентов [2, 9].
Выделяют 3 типа гипертрофии сердца: нормальную и индуцированную физической тренировкой и патологическими стимулами. Показано, что гипертрофия первых 2 типов регулируется главным образом через гормон роста/инсулиноподобный фактор роста путем сигнализации через путь фосфоинозитол-3-киназа/Akt. Патологическая гипертрофия запускается через путь Gq/фосфолипаза С, вызывающий увеличение цитозольного Са2+ и активацию протеинкиназы С [10].
При пролонгированном сердечно-сосудистом стрессе или патофизиологических стимулах миокард подвергается каскаду компенсаторных структурных изменений. Процесс протекает как континуум, обусловливающий ремоделирование структуры и различных функций миокарда. Ремоделирование желудочка представляет собой сложный процесс в результате взаимодействий между начальным повреждением или изменением нагрузки и множеством механических и нейрогуморальных факторов, которые способны модифицировать фенотип кардиомиоцитов и индуцировать изменения во внеклеточном матриксе. Последний является не статической структурой, а динамической компонентой, играющей фундаментальную роль в адаптации миокарда в ответ на патологический стресс и определяющей процесс ремоделирования миокарда. При ремоделировании миокарда структура
и состав внеклеточного матрикса подвергаются значительным изменениям [11, 12], влияя на взаимодействие, пролиферацию и миграцию клеток. Кардиомиоциты, фибробласты и эндотелиальные клетки дифференцированно экспрессируют и реагируют на определенные компоненты внеклеточного матрикса, которые вносят свой вклад в клеточную коммуникацию, влияя на функцию миокарда в целом.
Гипертрофия миоцитов, некроз и апоптоз клеток, интерстициальный фиброз и деградация коллагена являются главными особенностями ремоделирования желудочков. Каждый из этих компонентов ремоделирования вносит важный вклад в развитие и прогрессирование СН. На уровне самого желудочка ремоделирование связано с изменением геометрии, объема и массы. Хотя первоначально они являются компенсаторными в условиях нарастающего давления и объемной нагрузки, прогрессирующее ремоделирование ЛЖ в итоге предстает дизадаптивным процессом, содействуя усилению симптомов СН и приводя к худшему исходу [2, 8]. После острого ИМ прогрессирующая гипертрофия и ремоделирование сохранившегося миокарда могут быть с самого начала неблагоприятными. Ремоделирование после ИМ включает изменения в структуре коронарных сосудов, потерю миоцитов, гипертрофию оставшихся миоцитов и увеличение размера и числа немиоцитарных клеток. Все это в совокупности приводит к неоднородным изменениям геометрии стенки миокарда ЛЖ [13]. Безотносительно к первоначальному повреждению дилатация может стать самоподдерживающимся процессом ухудшения структуры и функции ЛЖ. Прогрессирующее увеличение камеры желудочка становится стимулом для дальнейшей гипертрофии и дилатации без дополнительного повреждения миокарда [2, 6, 8, 13].
Особенностью перепрограммирования экспрессии генов при гипертрофии сердца и СН является активация фетальных генов миокарда, которые кодируют белки, участвующие в сокращении, мобилизации Са2+ и метаболизме. В числе главных аномалий сократительных белков отмечаются изменение их содержания, переключение синтеза на другие изоформы, посттрансляционные
модификации и мутации [2]. Имеются доказательства, свидетельствующие о значительном уменьшении при СН синтеза мРНК α-тяжелой цепи миозина и возрастании фракции выброса при терапии β-адреноблокаторами, ассоциирующегося с нормализацией экспрессии ее мРНК. Кроме того, мутации гена α-тяжелой цепи миозина ассоциированы с гипертрофической кардиомиопатией, что демонстрирует критическую роль этого белка для нормальной функции миокарда. Отмечено снижение
экспрессии гена кальциевой АТФазы саркоплазматической сети при гипертрофии и СН, приводящее к снижению эффективности мобилизации Са2+. Экспрессия генов натрийуретических пептидов — предсердного (А) и мозгового (В), напротив, возрастает как проявление компенсаторного механизма для содействия натрийурезу и супрессии гипертрофии миоцитов. Такое транскрипционное репрограммирование коррелирует с ухудшением функции сердца, в то время как улучшение его функции в ответ на фармакотерапию или имплантацию вспомогательного устройства сопровождается нормализацией экспрессии генов [2, 13].
Изменения при СН охватывают практически все функциональные системы кардиомиоцитов, что порождает тот же вопрос, что и при нейродегенеративных заболеваниях: который из молекулярных механизмов в этом патогенетическом континууме является первичным и который — вторичным. Было бы рискованно пытаться представить эту пугающую сложность взаимодействий разных клеточных и молекулярных систем миокарда при СН даже многокомпонентной схемой. Эта осторожность особенно оправдана ввиду возрастающего числа сообщений о многоэтапном изменении транскрипции генома в экспериментальных моделях СН [14, 15]. Поэтому далее, как в этом, так и в последующих разделах статьи будут затронуты лишь наиболее резкие сдвиги в других основных функциональных системах клетки без упоминания конкретных молекулярных участников, поскольку даже в рамках огромной монографии [16] невозможно охватить нарастающий поток публикаций по СН.
В рамках эволюционирующей концепции патогенеза СН ремоделирование камер желудочков сердца обоснованно рассматривается в контексте ремоделирования метаболизма, энергообеспечения, потоков ионов, особенно мобилизации Са2+ в процессе мышечного сокращения и расслабления и других процессов, претерпевающих на начальных стадиях СН согласованные (точнее, взаимнорегулируемые) адаптивные изменения. В частности, изменение клеточного окружения (нагрузки, доступности субстратов, регуляторных факторов) влечет изменение сигнальной трансдукции и соответственно изменение метаболизма (транспорта глюкозы, окисления пирувата, β-окисления), метаболических сигналов (накопление метаболитов глюкозы и жирных кислот), активности факторов транскрипции и трансляции, экспрессии генов и белков (индукция фетальных белков и факторов роста) [17—22].
Метаболическая регуляция сложнейшим образом связана с функционированием сердца. Прогрессирование СН любой этиологии сопряжено с постепенным уменьшением активности дыхательных цепей митохондрий, ведя к сниженной способности образования аденозинтрифосфата (АТФ) и к вторичной дизрегуляции клеточных процессов, критических для насосной функции
(среди которых мобилизация Са2+ и сократительная способность) и приводящих к возрастанию энергетической потребности и сниженной функции миокарда. При этом пути генерирования АТФ не реагируют пропорционально динамическим флуктуациям при нагрузках и доставке топлива. Временна`я рамка регуляторных ответов на нагрузку или стресс затрагивает множество уровней,
среди которых аллостерический контроль ферментной активности через метаболические продукты, сигнальная трансдукция, регуляция генов, синтез белков. Если окисление жирных кислот и глюкозы в митохондриях поддерживает генерацию подавляющей части АТФ у здоровых и жирные кислоты являются предпочтительным субстратом в миокарде взрослого, обеспечивая до 70% общего количества АТФ, то при СН эти процессы оказываются резко нарушенными. В ходе патологического ремоделирования происходит сдвиг в способе утилизации энергии от окислительного к гликолитическому, что еще более способствует гибели кардиомиоцитов [17, 19—22].
Сдвиги в путях мобилизации Са2+, по-видимому, являются в числе особенно значимых в спектре патологических изменений при СН. Изменение в содержании и активности почти всех регулируемых Са2+ молекул вносят вклад в измененный гомеостаз Са2+ сердца. Однако маловероятно, чтобы изменение в содержании одной из регулируемых Са2+ молекул было бы ответственно за дефекты сократительной активности при СН. В то же время несомненно, что изменение в уровне сократительных белков, особенно тех, что связаны с тонкими нитями саркомера, являются важными компонентами патофизиологии выраженной СН. Данные у пациентов с семейной кардиомиопатией четко свидетельствуют о том, что единичные мутации, затрагивающие саркомерные белки, достаточны для индуцирования глубокого фенотипического изменения кардиомиоцитов [19].
Измененное образование и пространственно-временное распределение реактивных форм кислорода и азота создают окислительный и нитрозативный стрессы в самом сердце и в сосудистой системе, формируя их патологические фенотипы. Эти нарушения на интегративном системном уровне могут быть объяснены повреждающими эффектами в сигнальных элементах не только сердца и сосудистой
системы, но и крови, также участвующей в регулировании сердечно-сосудистого гомеостаза. S-нитрозилирование цистеина в белках является тем путем, через который NO модулирует различные клеточные процессы [17, 18]. Имеется множество доказательств того, что хроническая СН связана с нарушенной эндотелийзависимой вазодилататорной функцией периферической сосудистой системы. Хотя большинство этих нарушений может быть приписано сниженной биодоступности NO, точные механизмы, лежащие в основе этого, пока неизвестны. Несомненно, что нарушение функции эндотелия при хронической СН является мультифакторным и включает пониженную экспрессию эндотелиальной синтетазы NO, ограниченную доступность ее субстрата (аргинина), активацию ренин-ангиотензин-альдостероновой системы (РААС) и, вероятно, повышенную деградацию NO, вызванную инактивацией свободными радикалами [18, 23].
Нельзя не отметить также влияние на развитие СН активирования кардиальных источников РААС. Все ее компоненты, альтернативные пути формирования ангиотензина II — АТII (химазы, катепсина G) и сам ангиотензин II были идентифицированы в сердце человека. Существуют доказательства динамической экспрессии РААС в сердце человека при гипертрофии сердца и СН. Их регуляция, вероятно, обусловлена гемодинамической нагрузкой и инфильтрацией воспалительными клетками.
Новая область исследований СН связана с образованием АТII в сердце и сигнальными механизмами АТII, запускающими его интракринную продукцию в кардиомиоцитах, фибробластах и нейрональных ганглиях. В этих клетках экспрессия генов АТII и других компонентов РААС может увеличиваться при напряжении и электрической стимуляции. Ядерная мембрана, по-видимому, содержит рецептор АТII, который сопряжен с транскрипцией РНК. Новые данные подтверждают, что образование АТII может регулироваться не только растяжением, но и трансактивацией рецепторов, сопряженных с G-белками
и гормонами, и соответственно давать интракринно обусловленные клеточные эффекты, открывая альтернативные возможности их регулирования, помимо блокады РААС [24].
Возможны разные пути формирования патогенетического континуума при остром и хроническом варианте развития СН. Острое возникновение СН сопряжено с быстрой мобилизацией компенсаторных механизмов и реакциями на системном уровне. Исследование миокарда в экспериментальной модели СН от начала воздействия повреждающего фактора и в последующие за ним промежутки времени показало проявление глубоко различных паттернов экспрессии генов в каждой временной точке анализа со значительным преобладанием экспрессии генов какой-то одной функциональной предназначенности. В начале усилена экспрессия генов, ответственных за клеточный цикл, а в следующей временной точке превалирует экспрессия генов, ответственных за сигнальную трансдукцию, которая сменяется стадией преобладания экспрессии генов, ответственных за движение микротрубочек, и появлением мРНК с неизвестной функцией. За ней следует стадия преобладания экспрессии генов, ответственных за клеточную адгезию, и снижения общего уровня экспрессии при
нарастании в нем доли экспрессии генов неизвестной функции, которые значительно превалируют над остальными генами в последней проанализированной стадии [6]. При всей осторожности экстраполирования данных, регистрируемых в экспериментальных моделях СН, на патогенез СН у человека, выявление экспрессии генов с неизвестной функцией на поздних стадиях СН свидетельствует о том, что наши знания о патогенезе СН и по сей день далеки от полного его описания. Вполне вероятно, что на современном этапе развития биологии и медицины, часто характеризуемом как постгеномная эра, с его новым витком исследований возрастающей сложности, включающих системный анализ организма на основе достижений нанотехнологий, мы станем
свидетелями новых сюрпризов, преобразующих наши представления о патогенезе СН.
Старость и провоспалительный статус организма
В нашем меняющемся понимании СН как системного заболевания важно учитывать проявление ее преимущественно при старении организма и роль обеих систем (врожденной и адаптивной) иммунитета в формировании ее патогенетического континуума. Процесс старения сопряжен с прогрессирующим снижением структурной и функциональной стабильности организма, затрагивающим раньше и чаще всего сердечно-сосудистую систему, что усугубляется и сдвигом иммунной системы к провоспалительному и аутоиммунному статусу. Трудности разграничения между физиологическим и преждевременным старением, с одной стороны, и общность многих факторов (например, гипертонической болезни либо проявлений атеросклероза), ведущих к СН и преждевременному старению, с другой, побуждают признать возникающую СН как несомненный признак прогрессирования старения организма и исчерпания его адаптивных возможностей.
Каждый орган поддерживает свой клеточный гомеостаз в том временном ритме, который согласован с его функциональными потребностями и эволюционно запрограммированной стабильностью составляющих его клеточных элементов, внося свой вклад по обеспечению целостности организма. Обычно в детородном возрасте, когда обеспечивается воспроизведение человека как вида, нет проблем с функционированием мозга или сердца. Они возникают у молодых, как правило, при
генетических нарушениях и инфекционных поражениях, а в пожилом и старческом возрасте — в силу физической изношенности органов. Развитие цивилизации за короткий срок по меркам эволюции быстро увеличило продолжительность жизни человека, что фактически оказалось не зафиксированным в эволюционном опыте [12], и к тому же, как справедливо замечает R. Zinkernagel [25], в эволюционной перспективе нет преимущества защищать вид от заболевания, возникающего по прошествии детородного возраста. Это подводит к признанию того, что, вступая в период среднего возраста, человеческий организм не следует каким-то закрепленным эволюцией программам, как это происходит от момента его рождения до полового созревания, а пускается в «свободное плавание».
Однако неумолимая статистика свидетельствует о том, что это «плавание» вовсе не свободное и имеет свои модальности. В рамках рассматриваемой нами проблемы примечательно то, что биологические основы женского рода долговечнее мужского, и поскольку виновником наиболее распространенных заболеваний и наиболее высокой смертности человечества является сердечно-сосудистая система, то она среди других систем и органов наиболее уязвима. Рассматривая эти два факта во взаимосвязи, нельзя не отметить физиологически нормальные онтогенетические особенности клеточного гомеостаза сердца мужчин и женщин.
Зрелый миокард содержит 20—25% миоцитов и поддерживающую соединительную ткань. В процессе старения большинство компонентов миокарда подвергается структурным изменениям, проявляющимся потерей части миоцитов и гипертрофией оставшихся миоцитов. Последние часто полиплоидны. Одновременно отмечаются накопление коллагена, фиброз и отложение амилоида и липофусцина. Стареющие кардиомиоциты медленнее сокращаются и расслабляются. Обновление миоцитов снижается [26]. Если количество миоцитов в правом и левом желудочках сердца женщин до глубокой старости остается в основном постоянным, то у мужчин, начиная с 17 лет, ежегодно убывают 45∙106 и 19∙106 кардиомиоцитов в левом и правом желудочках соответственно. Потеря кардиомиоцитов у мужчин компенсируется гипертрофией остающихся клеток [27].
В процессе нормального старения сердце подвергается функциональным, морфологическим и клеточным изменениям. Хотя само по себе старение не ведет к проявлению СН, связанные с возрастом изменения, по-видимому, снижают порог проявления ее симптомов. У пациентов предрасположенность к СН, начало и степень ее прогрессирования резко различаются. Наличие известных факторов риска не может полностью объяснить эту вариабельность. Одна из объясняющих ее гипотез опирается на биологию теломеров [26].
Теломеры состоят из эволюционно консервативных повторяющихся нуклеотидных последовательностей и локализуются на концах хромосом. С каждым делением клетки в теломерах утрачивается 30—150 пар нуклеотидов, так как ДНК-полимераза не способна полностью реплицировать 3©- конец нити ДНК. С достижением критической длины теломера клетка утрачивает способность к делению и может стать дисфункциональной. В среднем это происходит спустя 50 удвоений (делений) клетки. Но не только длина самого теломера влияет на способность клеток к удвоению и их жизнеспособность, важна также и роль белков, связанных с ним. Длина теломеров
резко различается между индивидуумами и рассматривается в числе факторов, предрасполагающих к хронической СН, артериальной гипертонии, сахарному диабету, ИМ и активации РААС. Многие факторы окружающей среды способны влиять на износ теломеров, и наиболее важный из них — окислительный стресс. Биологическое старение сопряжено с уменьшением протяженности теломеров, чем пытаются объяснить резко варьирующий между индивидуумами порог клинических проявлений
СН на определенной стадии жизни. Накопление повреждений ДНК и истощение теломеров приводит к прогрессированию старения клеток и апоптозу, что в итоге снижает численность клеток и их функциональность в органе и приводит к дисфункции органа. Однако и по сей день длину теломеров можно рассматривать как эпифеномен или как следствие, но не как причину хронической СН.
Признано, что стволовые клетки, обеспечивающие клеточное обновление органа или ткани, аналогично соматическим клеткам обнаруживают с возрастом внутренние изменения, приводящие к их старению и утрате способности самообновляться и соответственно к нарушению функции органов [28—30].
Поскольку обычным исходом ИМ в случае выживания пациента являются рубцевание инфарктированной зоны и последующая прогрессирующая недостаточность сократительной функции сердца, то вплоть до недавнего времени полагали, что сердце не содержит резидентные стволовые клетки, способные восполнить утраченные кардиомиоциты. Сами же идеи относительно возможности обновления кардиомиоцитов воспринимались как фантастические. Однако в последние годы с помощью тонких методов было доказано, что не только сердце, но и мозг содержит резидентные стволовые клетки. Так как доля обновляющихся кардиомиоцитов очень мала, то большинство кардиологов не рассматривали эти данные как представляющие важное функциональное значение. Хотя и сегодня не всеми признается функциональная важность наличия стволовых клеток в сердце, и голоса скептиков и критиков еще не единичны, прочно устоявшиеся представления о невозможности обновления сердца взрослого человека начинают пересматриваться. Нетрудно подсчитать, что даже при медленных темпах образования новых кардиомиоцитов на протяжении человеческой жизни
клеточный состав сердца хотя бы раз обновляется полностью. Более того, обнаружена линия мышей, у которых практически полностью регенерирует сердце, пораженное инфарктом [31]. Показано, что в конечной стадии СН происходит репликация кардиомиоцитов, митотическая активность отмечается в периинфарктной области миокарда. Более того, недавно описано 4-кратное превышение числа резидентных прогениторных клеток в сердце при СН [28—30]. Хотя естественный уровень самообновления кардиомиоцитов при СН остается недостаточным, чтобы предотвратить ее развитие, исследователи не теряют надежд найти способ усиления этого процесса.
Возникающая несбалансированность между апоптозом поврежденных и образованием новых кардиомиоцитов, накопление стареющих кардиомиоцитов и стволовых клеток рассматриваются в числе факторов, определяющих процесс старения сердца и приводящих к его структурно-функциональной недостаточности [27, 32, 33]. Одна из особенностей старости — разрушительность, и у мужчин ее программа, затрагивающая сердце, начинает реализоваться рано — уже при достижении
половой зрелости, и соответственно у них намного раньше, чем у женщин, включаются компенсаторные механизмы поддержания надлежащей функциональной способности сердца и раньше наступает его изнашивание с известными последствиями. Поэтому возрастные сроки скринингового обследования мужчин для выявления бессимптомной СН должны быть более ранними с использованием биомаркеров, по крайней мере, компенсаторной стадии СН.
В организме надзирающей и разрушающей является иммунная система, усиливающая свой провоспалительный и аутоиммунный статус при старении. Вклад иммунной системы в развитие СН многосторонне исследовали за последние 15 лет [34, 35], и сейчас интерес к роли иммунного фактора при СН особенно усилился в контексте этиопатогенеза как определяющего медленное, хроническое и прогрессирующее ее развитие. При хронической СН происходит активация иммунной системы, которая
приводит к образованию и выделению провоспалительных цитокинов, активации комплемента, образованию антител, гиперэкспрессии главного комплекса гистосовместимости класса II, а также молекул адгезии, что может «увековечивать» воспалительное состояние. Цитокины являются ключевыми элементами в активации, распространении и усилении иммунного ответа. Они вовлечены
в рекрутирование клеток иммунной системы в зону воспаления, стимулирование клеточного деления, пролиферации и дифференциации. Гиперпродукция α-фактора некроза опухолей (α-ФНО), выделяемого, главным образом, активированными макрофагами и в меньшей мере моноцитами, лимфоцитами и нейтрофилами, вероятно, обостряет СН, вызывая повреждающие эффекты на периферии: скелетную миопатию, дисфункцию эндотелия и его апоптоз. Другие цитокины — интерлейкины (ИЛ) ИЛ-1, ИЛ-6, ИЛ-2, ИЛ-18 и интерферон-c также участвуют в патогенезе и прогрессировании СН, снижая сократимость миокарда, в то время как повышение скорости осаждения эритроцитов и С-реактивного белка, по-видимому, отражает уровень системного воспаления при СН.
Некоторые из иммунологических биомаркеров коррелируют с силой и прогнозом СН, воспроизводя ее развитие у пациентов без симптомов, изменяются под воздействием терапии СН и представляют потенциальные мишени для терапии в будущем. Сам миокард способен продуцировать различные цитокины и через них способствовать ремоделированию ЛЖ и оказывать отрицательный инотропный эффект. Это дает основание рассматривать хроническую СН как системное заболевание, а не только как проявление недостаточности насосной функции сердца с ее последствиями [34—37]. Поэтому в последние годы прилагаются активные усилия по использованию противовоспалительной терапии при СН [38—41].
Клиническое течение хронической СН трудно прогнозировать. При наличии сходной этиологии, фракции выброса желудочков и демографии понимание разнообразия ее исходов остается ограниченным. В числе факторов распространенности СН и разнообразия ее течения важная роль отводится иммунной системе, поскольку при неблагоприятном исходе прогрессивно повышаются
уровни цитокинов. Однако попытки блокировать развитие СН с помощью иммуномодулирующей терапии оказываются в основном малоэффективными, что, возможно, отражает недостаточность наших знаний о сложной роли иммунной системы в патогенезе хронической СН и упрощенного подхода к иммунотерапии. Возможно, что потенциально миокард, а не иммунная система, осуществляет контроль за клиренсом миоцитов посредством индуцирования гибели клеток и их удаления. Эта концепция находится в согласии с гипотезой P. Matzinger [42] о роли иммунной системы как реагирующей на повреждение или на субстанции, вызывающие повреждение, как и на субстанции, которые являются чужеродными. Эта концепция изначально была постулирована для инфекций и опухолей, однако хорошо совмещается и с современными представлениями о прогрессии болезни и вовлечении иммунной системы в патогенез хронической СН. Множество факторов, вызывающих хроническую СН через повреждение миоцитов, ведут к синдрому с идентичными характеристиками заболевания. Это могло бы происходить в целом или частично из-за потери аутотолерантности и распознания аутоантигенов посредством презентации их поврежденным миокардом клеткам иммунной системы [36].
В связи с неоднозначной оценкой разными исследователями роли одной и той же молекулы либо механизма (в том числе цитокинов) в патогенезе СН нельзя не заметить, что оно предопределено, по крайней мере, двумя факторами. Первым является свойственная сложным организмам вырожденность — выполнение одной и той же функции разными по своей структуре молекулами [43]. Вторым — гетерогенность (по стадии болезни, возрасту, полу, получаемой терапии и др.) среди пациентов.
Что же касается роли воспаления в сценарии запуска патогенетического континуума СН, то применительно к СН ишемического генеза, вызванного атеросклерозом коронарных артерий, он мог бы быть, например, следующим. Ограниченность сердца в способности использовать различные субстраты окисления обусловливает ранимость кардиомиоцитов даже при кратковременных локальных нарушениях коронарной гемодинамики. Обычно область ишемии неоднородна по развившимся в ней изменениям. Ее центральная часть состоит преимущественно из погибших клеток, а к ней прилегает зона функционально подавленных, но жизнеспособных клеток. Во время гемодинамического криза в ответ на повреждение миокарда активируются иммунная система и кардиофибробласты, запускается каскад изменений вокруг микроочага. В некротизированном микроочаге и вокруг него развивается воспалительная реакция, сопровождаемая усиленным образованием и выделением провоспалительных
цитокинов не только всеми типами клеток миокарда, но и инфильтрирующимися и скапливающимися в ишемическом очаге полиморфноядерными лейкоцитами, моноцитами/макрофагами. Повторяющиеся микроишемические эпизоды по своим прогрессирующим и накапливающимся повреждениям сердца не менее опасны, чем остро возникший крупноочаговый ИМ, поскольку способствуют хроническому поддержанию воспалительного процесса. К тому же первоначально возникшая воспалительная реакция и выделяемые при ее развитии провоспалительные цитокины из-за недостаточно сбалансированной регуляции могут вызвать гибель не только не утративших жизнеспособность депрессированных клеток вокруг ишемического очага, но и здоровых клеток, что порождает
новую волну выделения провоспалительных цитокинов и установление расползающегося самоподдерживающегося цикла: гибель клеток ↔ воспалительная реакция. Провоспалительный и проаутоиммунный статус иммунной системы в пожилом возрасте [44] служит фактором, благоприятствующим самораспространению такого цикла. Поэтому ишемическая болезнь сердца, сопутствуя гипертонической болезни, атеросклерозу или сахарному диабету и прогрессируя, при несбалансированной регуляции иммунной системы порождает накапливающиеся изменения миокарда, ведущие в итоге к клиническомупроявлению СН.
Биомаркеры СН
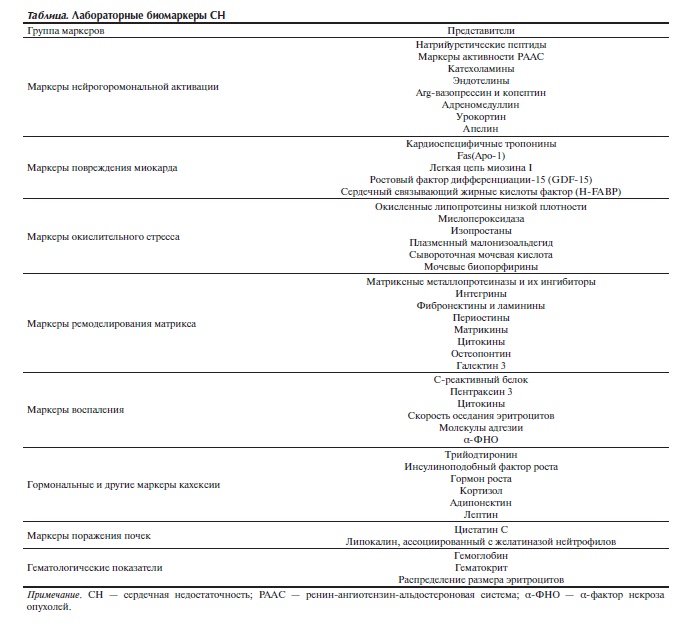
Проявление СН как системного синдрома позволяет использовать различные по своей природе его биомаркеры. Их нарастающая численность свидетельствует о сложности патогенеза СН и о необходимости интеграции их в общую концепцию патогенеза СН, используя системные подходы. Априорно в числе биомаркеров могут быть молекулы, выделяемые самим сердцем при физиологических и патологических состояниях, и молекулы, продуцируемые в других интегрирующих системах организма в ответ на возникающее и затем прогрессирующее нарушение сердечно-сосудистой системы. Среди них показатели повреждения самих кардиомиоцитов, изменения нейрогуморальной активации, ремоделирования внеклеточного матрикса, дисфункции почек и др. (см. таблицу). Отметим лишь некоторые из них. Наибольшее признание в качестве маркера СН, в том числе ее досимптоматической стадии, получил мозговой натрийуретический пептид, синтезируемый желудочками сердца. При его содержании выше 80—100 пкг/мл иммунологические тесты обеспечивают весьма высокие показатели специфичности и чувствительности выявления у пациентов СН [45]. Другими кандидатами в маркеры СН могут служить кардиоспецифичные тропонины Т и I, белок, связывающий жирные кислоты, и легкая миозиновая цепь. Как и мозговой натрийуретический пептид, они происходят из миокарда и могут рассматриваться как специфические маркеры. Их концентрация возрастает в отсутствие ишемических приступов у пациентов с тяжелой формой
СН [46].
Таблица. Лабораторные биомаркеры СН.
В числе новых биомаркеров СН следует отметить белок галектин 3. Он является представителем семейства лектинов, распознающих углеводные компоненты и играющих важную роль при воспалении, иммунитете и раке. При СН он отражает активацию фибробластов и макрофагов, связанную с ремоделированием сердца. Увеличение концентрации галектина 3 служит индикатором развития и прогрессии СН и наступления декомпенсации, предвещая летальный исход [47].
Другим биомаркером со сходной прогностической характеристикой СН является копептин — С-концевой фрагмент (из 39 аминокислот) предшественника Arg-вазопрессина. Он зеркально отражает косекрецию вазопрессина, прогностический потенциал которого хорошо известен применительно к СН, и более стабилен, чем объясняется предпочтительное его использование в кар-
диологии. У пациентов с хронической СН при высоких уровнях копептина отмечен значительно худший долгосрочный прогноз. Копептин рассматривается в качестве более сильного предиктора летальности у пациентов с ИМ, чем мозговой натрийуретический пептид и его N-концевой предшественник [48].
Кардиотонические стероиды млекопитающих (карденолиды) представляют семейство эндогенных соединений, аналогичных по структуре дигоксину и оубаину из растений и участвующих в поддержании сердечно-сосудистого гомеостаза. Синтез и секреция карденолидов осуществляются главным образом надпочечниками и гипоталамусом. В надпочечнике синтезируются и дигоксин-подобные и оубаин-подобный иммунореактивные факторы, а в гипоталамусе — только оубаин-подобный фактор. Хотя концентрация карденолидов увеличивается при различных клинических состояниях, сопровождающихся гиперволемией, почти двукратное возрастание их концентрации по сравнению с нормой может служить основанием для диагноза бессимптомной СН [45].
СН, как отмечалось, сопровождается увеличением концентрации различных цитокинов, с которыми связывают проявления ее многих клинических признаков. Источниками увеличения концентрации цитокинов при СН служит не только иммунная система, но и сам миокард. Провоспалительные цитокины, α-ФНО, ИЛ-6 и С-реактивный белок чаще всего используются в качестве биомаркеров СН. Недостатком цитокинов в качестве маркеров является заметное их возрастание преимущественно в поздних стадиях СН [45, 49].
Хотя клиницистам нелегко воспринимать математические подходы к оценке вариабельности ритма сердечных сокращений, они (особенно нелинейные методы) позволяют уловить тонкие изменения разных показателей ритма. Их неинвазивность, быстрота анализа и возможность многократного мониторирования позволяют прослеживать динамику состояния сократительной способности миокарда при СН [50].
Рациональным представляется классифицирование биомаркеров по наилучшему отображению ими разных аспектов СН, например, диагностики, определения стадии, прогноза и терапевтического мониторирования и т.д. Помимо хорошей диагностической и прогностической точности общими критериями для каждого маркера следует считать приемлемость их для пациента, стабильность
in vivo и in vitro, аналитическую чувствительность, воспроизводимость и точность, возможность автоматического определения, международную стандартизацию, низкую стоимость, невысокую биологическую вариабельность, наличие данных, связанных с особенностями их величин в зависимости от пола, возраста или этнической принадлежности [51—54].
В аспекте существующей в организме динамичности функциональной связности между молекулами и необратимого по существу прогрессирования СН становится очевидным, что один и тот же биохимический маркер не может быть идеальным, являясь одновременно кардиоспецифическим, прогностическим и не зависимым от других маркеров, отражая прогрессирование СН, как и ее реакцию на терапию, тем самым позволяя диагностирование не только ранних стадий СН, и быть надежным индикатором ее различных стадий. Об этом свидетельствует прежде всего само множество предложенных маркеров СН, отражающих состояние различных систем организма, функция которых подвергается наиболее сильным нарушениям. Эволюционирование концепции патогенеза СН свидетельствует о невозможности свести его к какому-то одному механизму и, следовательно, объяснить какой-то одной истиной. Согласно теоретикосистемному подходу поведение системы обусловливается вкладом всех ее компонент, а наибольшее влияние на изменение ее оказывают управляющие параметры, или системные переменные [55].
В развивающемся патогенетическом континууме СН не все изменения множества молекулярных и физиологических механизмов следует воспринимать как патологические на определенных его стадиях, поскольку изначально они являются адаптивными и направлены на компенсацию недостаточной функции, а конкретно — для поддержания сократительной способности миокарда и его насосной функции. Необходима осторожность в интерпретации роли и вклада каждого механизма в патогенез СН и в поисках новых маркеров и терапевтических мишеней. К примеру, вначале воспалительная
реакция в ответ на повреждение или инфекционный патоген рассматривается как полезная, но при задержке разрешения воспалительной реакции ее деструктивные проявления намного превосходят первоначально возникшее повреждение.
Генетические маркеры
Последнее десятилетие ознаменовалось расширением спектра биомаркеров СН за счет изучения новых походов, связанных с характеристикой генома: геномикой и транскриптомикой, характеризующих соответственно нуклеотидную последовательность генома и спектр транскрибируемых с него мРНК. Для анализа профиля геномики пациента может быть использована ДНК из любой клетки организма в любой момент его жизни, в то время как профили исследования экспрессии генов, будь то мРНК или
белки, связаны со специфическими обстоятельствами. Они существенно варьируют от органа к органу, момента жизни, стресса, лечения и других факторов.
Наиболее успешными исследования геномики оказались в случае первичных гипертрофических карди-
омиопатий. Выявлено, что они могут быть обусловлены 450 различными мутациями и 50—70% их обусловлены вовлечением одного гена и связаны с саркомерными белками кардиомиоцитов, причем нередкой оказывается ситуация, когда семья характеризуется своей «частной мутацией». Множество различных мутаций описано и при дилатационной кардиомиопатии. Причем некоторые мутации в одних и тех же генах могут вызывать либо гипертрофическую, либо дилатационную кардиомиопатию. Например, мутации в кардиоспецифичном тропонине Т, титине, связанном с миозином белке С и тропо-
миозине. Эти наблюдения подчеркивают, что изменения в ДНК не полностью определяют конкретный фенотип, поскольку эпигенетические факторы и взаимодействия генов могут изменять экспрессию первичной мутации [56, 57].
Более распространенными, чем мутации, являются вариации в последовательности нуклеотидов в ДНК, которые способны вносить вклад в чувствительность к заболеванию. Большинство этих вариаций представляют собой так называемые единичные нуклеотидные полиморфизмы, которые обусловливают различие индивидуумов по одной паре оснований. В отдельных популяциях некоторые из единичных нуклеотидных полиморфизмов встречаются с повышенной частотой. Они могут локализоваться в участках ДНК (экзонах), кодирующих белки, изменяя их аминокислотную последовательность, либо в некодирующих областях (интронах), влияя на сплайсинг мРНК и связывание регуляторных факторов. Важно, что эти вариации в последовательности ДНК изменяют чувствительность и реакцию индивидуума на патогены, химические вещества, в том числе лекарственные препараты. В этом отношении весьма показателен пример с корином в кардиомиоцитах, представляющим собой трансмембранную сериновую протеазу, расщепляющую предшественник натрийуретического пептида на его биологически активные формы. Наследование минорного аллеля корина связано с предрасположенностью к артериальной гипертонии и гипертрофии миокарда [58].
Эффекты вариаций генов могут проявляться очень сложным образом, приводя к сверхэкспрессии или недостаточности множества молекул. К примеру, экспериментальная модификация на мышах сверхэкспрессии кардиоспецифичного α-ФНО приводила к дисрегуляции более 1000 генов даже на стадии компенсаторной гипертрофии в отсутствие значительной дисфункции сокращения миокарда или дилатации ЛЖ [59]. Можно полагать, что не только изменения в гене, приводящие к сверхэкспресии той или иной молекулы, но и повышенная экспрессия тех или иных молекул под влиянием воздействия внешних факторов могут приводить к множественным и различным адаптациям на молекулярном уровне. Степень проявления изменений в геноме зависит от возраста, конкретной изоформы соответствующего белка, эпигенетических наслоений, вариации транскрипционного сплайсинга, профиля микроРНК (малых регуляторных РНК) [56, 60].
Очевидно, что, будучи финальным проявлением большинства сердечно-сосудистых заболеваний, СН может инициализироваться несколькими патофизиологическими процессами, вовлекая разные системы генов и регуляторных факторов. В случае семейной кардиомиопатии либо аритмии анализ генома позволяет выявить сеть генов-кандидатов или гена с мутацией, ассоциированных с последующим патогенезом СН.
Перспективы в изучении патогенеза СН
Достижения в фундаментальных науках и научных технологиях открывают новые возможности изучения патогенеза ранних стадий СН и вклада в ее прогрессирование иммунной системы, а также для диагностики, прогноза и оценки терапии СН на основе анализа динамики множества различных биомаркеров. Признано, что использование лишь генетических маркеров также имеет ограничения, поскольку большинство сердечно-сосудистых заболеваний имеют мультифакторную этиологию,
складывающуюся из сочетания генетических факторов с факторами окружающей среды, поведенческими особенностями и возникающими эпигенетическими наслоениями [56].
Эпигенетика представляет собой «экстраслои» контроля над геномом и определяется как модификации ДНК (без изменения самой последовательности нуклеотидов) и связанных с нею факторов, которые несут информацию и наследуются. Ее механизмы реализуются через метилирование (только цитозина в динуклеотиде CpG) ДНК, модификации (метилирование, ацетилирование или фофсорилирование) гистонов в нуклеосомах хроматина и геномного импринтинга. Метилирование ДНК и модификации гистонов — взаимосвязанные феномены, которые вместе определяют экспрессию генов. Патологические эпигенетические изменения ныне рассматриваются как мощные эквиваленты мутаций и хромосомных изменений [61, 62]. Пластичность эпигеномного уровня регуляции клетки объясняет уязвимость нашего организма при влиянии окружающей среды, питания, особенностей профессии, вредных привычек. Он включает участие таких глобальных регуляторов, как некодирующие РНК, в том числе микроРНК [60]. Не отрицая
важность исследования всего генома при СН и связанных с ней мутировавших генов, следует признать, что они будут явно недостаточными без изучения эпигенома на молекулярном уровне, охватывающем анализ влияния глобальных изменений, связанных с гипометилированием, гиперметилированием ДНК и изменениями хромосомальных белков. Например, показано, что стрессовые реакции сердца контролируют генную экспрессию изменением активности хроматин-ремоделирующим ферментом деацетилазой гистонов. Ее замечательная особенность — челночные переходы из ядра в цитоплазму
и обратно в зависимости от функционального состояния клетки. Известны 3 класса этого фермента. К отдельным их представителям были синтезированы ингибиторы, и при проверке их фармакотерапевтической активности не обошлось без сюрпризов. Оказалось, что ингибиторы деацетилазы гистонов сильно репрессируют агонистзависимую гипертрофию сердца по способу, который коррелирует с повышенным ацетилированием гистонов. Это парадоксально, поскольку деацетилаза класса II блокирует гипертрофию, и предполагалось, что ингибиторы деацетилазы гистонов нейтрализуют эту депрессивную функцию. Хотя механизм действия ингибиторов деацетилазы гистонов остается неясным, после нескольких десятилетий усилий в попытках модифицировать функцию сердца через традиционные терапевтические цели стали, наконец, вырисовываться возможности влиять на него, изменяя транскрипцию генома [2].
Ныне патогенетический континуум СН дополняется описанием белкового спектра и метаболизма организма (именуемые соответственно «протеомика» и «метаболомика»), что позволяет расширить описание функционирование организма как системы в виде сетевой структуры, более сложной, чем известные в биохимии метаболические сети. Сравнение профилей геномики, транскриптомики, протеомики и метаболомики пациентов и здоровых субъектов и соответственно сравнение этих профилей между пациентами позволяет «привязать» СН к определенным изменениям в этих профилях и дифференцировать лиц, отвечающих на терапию определенными препаратами, от не отвечающих на лечение, т.е. шире и глубже реализовать первоначальную идею индивидуальной фармакогенетики в терапии [63].
Интеграция этого потока информации, характеризующей наиболее существенные аспекты клеточной организации и физиологии организма, возможна в рамках системной биологии (биомедомики) [64]. Она делает свои первые шаги. С накоплением данных, объем которых еще более возрастет с бурно развивающимися нанотехнологиями, потребуется привлечение математического моделирования, теории сетей и статистических методов. Это поможет раскрытию патогенетического континуума СН
и обеспечит индивидуализированную терапию. Можно надеяться, что восприятие новой сложной диагностической информации будет облегчено созданием специализированных компьютерных пользовательских интерфейсов, которые сделают ее понятной для клинициста.



{kind=link}
{kind=link}