
Синдром удлиненного интервала QT (СУИQT) — одно из наиболее опасных заболеваний с риском развития внезапной аритмической смерти. Впервые описанный в 1957 г. норвежскими врачами F. Jervell и А. Lange-Nilsen, долгое время он существовал в двух вариантах — как синдром Джервела—Ланге-Нильсена (удлинение интервала QT, синкопе и внезапная смерть — ВС в семье у больных с врожденной глухотой) и как синдром Романо—Уорда (те же проявления у больных без глухоты) [1—3]. Однако успехи молекулярной генетики в аритмологии позволили выявить более 10 молекулярно-генетических вариантов заболевания, отличающихся не только по механизму нарушения функции ионных каналов кардиомиоцита (все варианты СУИQT относятся к классу каналопатий), но и нередко требуют генспецифической коррекции терапии.
В настоящем сообщении представлен клинический случай редкого варианта СУИQT — синдрома Андерсена—Тавила (САТ), относящегося к 7-му варианту СУИQT.
Описание случая
В ЦСССА ФМБА России обратилась женщина 34 лет с жалобами на приступы потери сознания. Первое предобморочное состояние возникло в 2009 г., во время вождения автомобиля, когда на фоне стрессовой ситуации на дороге больная стала терять сознание, захрипела. При последующем обследовании на электрокардиограмме (ЭКГ) была выявлена аритмия (желудочковая экстрасистолия по типу бигеминии, полиморфная тахикардия; рис. 1).
Рисунок 1.ЭКГ больной У., 32 года, после первого приступа потери сознания.
ЧСС 55 уд/мин, горизонтальное положение ЭОС, желудочковая экстрасистолия с отклонением ЭОС вправо/вниз; интервал PR 0,18 с, ширина комплекса QRS 60 мс, продолжительность интервала QT в отведении V5 500 мс. В грудных отведениях полиморфная желудочковая тахикардия (3, 4 и 5-й комплекс QRS) со средней частотой 133 уд/мин с последующей желудочковой экстрасистолией. Здесь и на рис. 2,4,5: ЧСС — частота сердечных сокращений; ЭОС — электрическая ось сердца.
После консультации в одной из московских клиник сделано заключение об отсутствии структурных и органических поражений сердца, назначен этацизин 50 мг 3 раза в сутки, на фоне которого количество аритмий значительно сократилось (рис. 2).
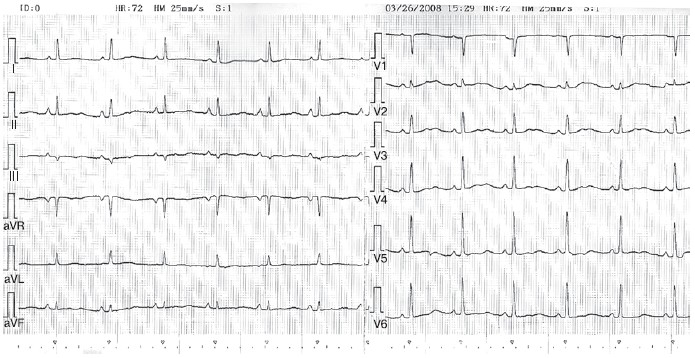
Рисунок 2.ЭКГ больной У., 32 года, на фоне приема этацизина.
ЧСС 72 уд/мин, горизонтальное положение ЭОС, интервал PR 0,18 c, ширина комплекса QRS 80 мс, бифазные зубцы Т в грудных отведениях, продолжительность интервала QT 540 мс, QTc — 596 мс.
Этацизин больная получала в течение 2 лет, после чего самостоятельно отменила. Второй обморок произошел летом 2011 г. — во время прогулки с собакой больная упала и потеряла сознание. Детали не помнит, домой привели очевидцы. Следующий обморок был в конце сентября 2011 г. дома (сидела на корточках у шкафа, перебирала вещи, внезапно кратковременно потеряла сознание), при этом мать отметила неправильное дыхание и подергивание мышц лица.
При осмотре в ЦСССА: правильное телосложение, кожа смуглая, масса тела 65 кг, рост 168 см. Обращают внимание стигмы дизэмбриогенеза: низко посаженные уши, скошенный подбородок, короткие пальцы (брахидактилия), умеренно выраженная клинодактилия V пальца на левой руке.
Границы сердца не расширены, тоны сердца ясные ритмичные, шумы не выслушиваются, частота сердечных сокращений (ЧСС) 72 уд/мин, артериальное давление (АД) 100/60 мм рт.ст. Больная отмечает периодически сниженное АД до 90/60 мм рт.ст. Семейный анамнез по случаям ВС, со слов больной и ее матери, не отягощен. При анализе ЭКГ родственников отмечена атриовентрикулярная блокада I степени у отца, интервал QT у всех родственников в пределах нормы.
Стандартные лабораторные показатели крови, мочи, электролиты (в том числе калий) у больной были в норме. Результаты исследований ЭКГ представлены на рис. 3. У больной отмечалась (рис. 3, а) частая ранняя желудочковая бигеминия в положении лежа (подсчет интервала QT и его производных из-за постоянной бигеминии невозможен), регистрировался залп двунаправленной желудочковой тахикардии (ЖТ) в отведениях II и V5 (рис. 3, б). В ортостазе аритмия исчезала, продолжительность интервала QT в отведении V5 составляла 540 мс, QTc — 596 мс (см. рис. 3, в).
Рисунок 3.ЭКГ больной У., 32 года, при обследовании в ЦСССА.
а — в положении лежа: ЧСС 70 уд/мин, интервал PR 0,16 с, ширина комплекса QRS 74 мс, подсчет продолжительности интервала QT из-за постоянной бигеминии невозможен; б — в положении стоя: ЧСС 75 уд/мин, интервал PR 0,16 с, ширина комплекса QRS 70 мс, продолжительность интервала QT в отведении V5 540 мс, QTc — 596 мс; в — разнонаправленная желудочковая экстрасистолия в стандартных отведениях (3-й комплекс QRS в положении лежа с отклонением ЭОС влево/вверх, 2-й комплекс QRS в положении стоя с отклонением ЭОС вправо/вниз), последние 3 комплекса QRS в положении стоя, с морфологией блокады правой ножки в грудных отведениях и двунаправленная желудочковая тахикардия во II отведении (последние 3 комплекса в отведении II записи ритма).
По результатам ЭКГ высокого разрешения поздние потенциалы желудочков не выявлены. При эхокардиографии патология не выявлена, полости и сократительная способность миокарда в норме. По результатам холтеровского мониторирования (ХМ) на аппарате MARS, GE 7.2 на протяжении всех суток регистрировалась тенденция к брадикардии (средняя суточная ЧСС 60 уд/мин при норме 70—80 уд/мин), правильный циркадный профиль ритма (циркадный индекс 1,27). Зарегистрированы 1806 одиночных и 829 парных полиморфных желудочковых экстрасистол, периоды би-, тригеминии, 128 эпизодов пробежки двунаправленной ЖТ (рис. 4), продолжительностью 3—11 комплексов с ЧСС 108—172 уд/мин, максимальное количество которых регистрировалось в период с 23.00 до 02.00 часов во время занятий домашними делами.
Рисунок 4. Полиморфная двунаправленная желудочковая тахикардия с ЧСС 158-164 уд/мин при холтеровском мониторировании.
«Плотность» желудочковой аритмии (соотношение числа желудочковых тахиаритмических комплексов к синусовым) составила 23%. Минимальная ЧСС составляла 36 уд/мин, при которой при ручном измерении интервал QT составил 672 мс (норма до 530 мс). Макровольтная (видимая) альтернация волны Т не зарегистрирована. Оценки частотной адаптации (динамики QT) и микровольтной альтернации волны Т (ТВА) оказались невозможными из-за полиморфной морфологии зубца Т и расхождения кривых при анализе ТВА. Турбулентность ритма сердца после экстрасистол не превышала норму.
С учетом характерной клинической картины (синкопальные состояния, периодически на фоне стресса), данных ЭКГ и ХМ (выраженное удлинение интервала QT на ЭКГ и при ХМ, залпы полиморфной двунаправленной ЖТ), типичные фенотипические изменения (низко посаженные уши, скошенный подбородок, брахидактилия и клинодактилия V пальца) больной поставлен диагноз: синдром удлиненного интервала QT. Синдром Андерсена—Тавила, 7-й молекулярно-генетический вариант? Больная была направлена в Медико-генетический научный центр РАМН для поиска мутаций в гене KSNJ2, типичных для данного варианта, которые методом прямого автоматического секвенирования выявлены не были. Больные с отрицательным генетическим анализом относятся ко второй группе больных с САТ [4—10].
Данные анамнеза, клинической картины и результатов обследования послужили основанием для назначения антиаритмическеой терапии — коргард (надолол) 1 мг/кг утром. На фоне данной терапии через 2 нед при контрольном обследовании на ЭКГ покоя регистрировалась синусовая брадикардия с ЧСС 62 уд/мин, продолжительность интервала QTc 490 мс лежа и 548 мс в ортостазе. Плотность желудочковой аритмии при ХМ снизилась в 4 раза — с 23 до 6% (полностью исчезли залпы ЖТ, сохранялись 5538 одиночных и 5 парных полиморфных желудочковых экстрасистол). Максимальная частота желудочковой аритмии с 22.30 до 00 ч (во время прогулки с собакой, подготовки ко сну), минимальная (до 0 в час) в период с 05.00 до 06.00 ч и с 08.00 до 09.00 ч утра. Минимальная ЧСС 46 уд/мин, интервал QT при которой составил 528 мс (666 мс QT + зубцы U). При контрольном обследовании через 2 нед количество желудочковых сокращений возросло до 15% (11 171 одиночных экстрасистол, 9 парных, в том числе двунаправленных), спонтанные паузы ритма достигли 2121 мс. С учетом появления признаков брадиаритмии на фоне приема β-адреноблокаторов, увеличения количества экстрасистол решено добавить к терапии препарат 1С класса этацизин 50 мг/сут на основании его предыдущей высокой антиаритмической активности у больной и имеющихся данных об эффективности препаратов данного класса у больных с САТ, но не отменять коргард, так как β-адреноблокаторы являются единственным классом препаратов, эффективно снижающих риск развития угрожающих жизни аритмий и ВС в этой группе больных. При контрольном ХМ через 7 дней плотность желудочковой тахиаритмии снизилась до 4% (3456 единичных желудочковых экстрасистол), паузы ритма уменьшились до 1736 мс (норма при ХМ), минимальная ЧСС увеличилась от 38 до 44 уд/мин, но сохранялось удлинение интервала QT до 640 мс.
Дальнейшее наблюдение больной предполагает динамическое обследование, включающее регулярное проведение ЭКГ и ХМ, расширение семейного клинико-электрокардиографического и генетического исследования, контроль уровня электролитов крови (прежде всего калия).
При возникновении синкопе на фоне приема коргарда — имплантация кардиовертера-дефибриллятора.
САТ — довольно редкое заболевание, сочетающее в себе неврологические, кардиологические и генетические механизмы и клинические проявления. Точная частота развития САТ неизвестна, к настоящему времени описаны около 100 больных по всему миру. Типичной клинической триадой САТ является: 1) наличие периодических гипо-, гипер- или нормокалиемических параличей (65, 20 и 15% соответственно [4]); 2) стигмы дисэмбриогенеза; 3) удлинение интервала QT на ЭКГ и сопутствующие ему желудочковые тахиаритмии.
Впервые 2 случая периодических параличей в сочетании с аритмиями описаны R. Klein и соавт. [5] в 1963 г. Однако возможная связь между данными состояниями не предполагалась. Только в 1971 г. Е. Andersen и соавт. [6] описали характерную триаду признаков у 8-летнего мальчика, и почти четверть века спустя 10 ранее описанных случаев были расширены до 14 пациентов. Первоначально синдром назывался по имени Андерсена [6], впоследствии к названию добавили имя Rabi Tawil, определяя вклад последнего в изучение механизмов заболевания [4, 7].
Заболевание наследуется по аутосомно-доминантному типу. По генетическим механизмам различают два типа САТ. Около 60% больных имеют мутации в гене KCNJ2, кодирующем α-субъединицы калиевых каналов Kir2.1 (1-й тип синдрома) [8, 9]. Движение ионов калия через эти каналы очень важно для поддержания нормальной функции как скелетных мышц, так и сердечной мышцы. Молекулярно-генетический механизм остальных 40% KCNJ2-негативных случаев, обозначенных как 2-й тип синдрома (как в нашем случае), остается неизвестен.
Периодические параличи могут быть первым признаком заболевания, САТ необходимо дифференцировать от других видов периодических параличей. Он составляет примерно 10% (1:500000) случаев всех периодических параличей [4, 11, 12]. Прежде всего, необходимо исключение вторичных причин развития периодических параличей (нарушения функции щитовидной железы, почек, надпочечников), устранение которых может быть эффективным методом лечения. Вторым шагом является определение уровня калия в сыворотке крови, который после приступов снижается. Электромиографические исследования по методике, разработанной E. Fournier [13], активно используются у больных с периодическими параличами для выявления миотонии, но у больных с САТ к настоящему времени не выявлено миотонии ни в одном исследовании с использованием данной методики. В нашем случае частых проявлений периодических параличей не отмечалось, однако мать больной периодически отмечала у нее непроизвольные мимические подергивания мышц лица. Уровень калия в биохимических анализах крови был в норме. Специфические для данного синдрома дисморфические изменения у нашей пациентки заключались в типичном паттерне лица (низко посаженные уши, скошенный подбородок, широко расставленные глаза), рук (брахидактилия и умеренно выраженная клинодактилия V пальца). Не отмечено синдактилии пальцев, но данный признак у больных с СУИQT более типичен для синдрома Тимотти (8-го молекулярно-генетического варианта СУИQT) — крайне злокачественной формы заболевания, при которой без ранней диагностики и лечения больные обычно внезапно умирают, не дожив до 3 лет [14].
Кардиальные проявления САТ включают удлинение интервала QT (у 50% больных), ЖТ (у 84% больных), из них двунаправленная ЖТ, наиболее частая аритмия, встречается у 32% пациентов, и остановка сердца регистрируется у 10% [9]. Полиморфная двунаправленная ЖТ также типична для другой каналопатии — катехоламинергической желудочковой тахикардии (КЖТ), при которой также отсутствуют очевидные признаки поражения миокарда, но нет удлинения интервала QT. Синкопе происходят исключительно на фоне повышенной физической и/или эмоциональной нагрузки, преимущественно в дневные часы [15, 16]. Молекулярно-генетической основой КЖТ являются мутации в гене RyR2, кодирующем рианодиновый рецептор, который способствует высвобождению кальция из саркоплазматической сети, и гене CASQ2, кодирующем кальсевквестрин.
Для всех больных с КЖТ типична выраженная синусовая брадикардия на ЭКГ. В ряде случаев отмечается типичный паттерн, заключающийся в синусовой брадикардии, укорочении PR интервала, увеличении циркадного индекса более 1,45 при норме 1,24—1,38 (признак повышенной чувствительности ритма сердца к катехоламинам) при ХМ [17—20]. В нашем случае при первичном обследовании, проведенном без приема препаратов, ЧСС по данным ЭКГ и ХМ соответствовала возрастной норме, а циркадный индекс составил 1,27. Основными дифференциально-диагностическими признаками разделения САТ и КЖТ явились удлинение интервала QT, нетипичность клинической картины приступов потери сознания (однократно в покое) и их поздний дебют (в 34 года). При типичной КЖТ приступы обычно начинаются в 5—7 лет, а к периоду совершеннолетия практически у всех больных наблюдаются синкопе. Кроме того, при КЖТ отсутствуют стигмы дизэмбриогенеза, типичные при САТ.
Иногда у больных с САТ выделяют специфическую картину ЭКГ, включающую паттерн ЭКГ, который состоит из удлинения интервала QT и бифазного или специфического зубца Т—U. В исследовании L. Zhang и соавт. [21] описан специфический паттерн Т—U зубца Т, который с чувствительностью 84% и специфичностью 97% наблюдался у больных с САТ, носителей KSNJ2-мутаций (1-й тип), в отличие от больных с отрицательным результатом генетического анализа.
Лечение САТ является одной из наиболее трудных задач вследствие редкости заболевания, полиморфизма клинических проявлений и малоизученных звеньев патогенеза. В лечении периодических параличей, в том числе при САТ [4, 6], эффективно используются ингибиторы карбоангидразы, такие как ацетазоламин (250—1500 мг/сут) и дихлорфенамид (20—200 мг/сут). Однако у 10% пациентов они могут вызвать нефролитиаз, и в таких случаях необходимо применение калийсберегающих диуретиков — спиронолактона (25—100 мг/сут) или триамтерена (25—100 мг/сут). Возможно включение в лечение калия [22]. Лечение аритмий во многом зависит от клинической картины. Частые комплексные желудочковые экстрасистолы реже трансформируются в угрожающие жизни аритмии, чем при других вариантах CУИQT [9, 15, 23, 24]. Однако постоянная длительная антиаритмическая терапия необходима для предотвращения угрожающей жизни тахикардии torsade de pointes, которая может привести к ВС больного. Как и при других вариантах СУИQT, требуется объяснить пациенту необходимость изменения образа жизни с исключением резких физических или эмоциональных нагрузок, приема препаратов, удлиняющих интервал QT. Стандартной терапией является назначение β-адреноблокаторов в дозе 2—3 мг/кг, эффект которых определяется при стресс-тесте, максимальная ЧСС при этом не должна превышать 130 уд/мин. В некоторых наблюдениях у больных с САТ отмечалась эффективность антагонистов кальция (амлодипин, нифедипин), изолированно или в комбинации с β-адреноблокаторами. Неясна роль препаратов I класса в лечении САТ. Многие из них удлиняют интервал QT, однако отмечены случаи успешного устранения желудочковых аритмий при их использовании. В работе D. Fox и соавт. [25] описывается случай наблюдения за 54-летним больным с типичными клинико-электрокардиографическими проявлениями САТ, частой желудочковой экстрасистолией (15 611 в сутки), залпами мономорфной и двунаправленной ЖТ. Больному был назначен бисопролол в суточной дозе 5 мг в комбинации с препаратами калия. Однако лечение эффекта не дало и бисопролол был заменен флекаинидом (100 мг), на фоне терапии которым отмечены выраженная редукция желудочковой экстрасистолии (до 268 в сутки), отсутствие залпов ЖТ. В нескольких других наблюдениях также отмечены эффективность и безопасность применения флекаинида у больных с САТ [26, 27].
Как известно, при САТ ионный дефект поражает α-субъединицу калиевого тока выпрямления Ik1, что приводит к удлинению терминальной фазы сердечного потенциала действия, продуцирующего раннюю постдеполяризацию. Депрессия канала Ik1 также приводит к перегрузке ионами кальция саркоплазматической сети, что является причиной поздней постдеполяризации, приводящей к триггерной активности кардиомиоцита. Механизм антиаритмического действия флекаинида (препарат 1С класса, блокатор натриевых каналов) у больных с САТ пока непонятен; возможно, взаимодействие калиевых и натриевых каналов в условиях перегрузки ионами кальция снижает триггерную активность кардиомиоцитов. У представленной больной хороший антиаритмический эффект был получен ранее при использовании этацизина — отечественного препарата того же класса и механизма действия, что и флекаинид. Поэтому при нарастании брадиаритмии на фоне приема коргарда мы добавили к терапии этацизин. Однако мы решили оставить в терапии β-адреноблокатор коргард, так как эффективность антиаритмического действия препаратов I класса, как показало исследование CAST I, не всегда совпадает с их возможностью предупреждать ВС [28]. Малое число описанных больных с САТ не позволяет пока доказательно обсуждать вопрос об эффективности проводимой нами комбинированной антиаритмической терапии, однако по мере накопления наблюдаемых случаев, продолжительности катамнестического наблюдения эта проблема будет проясняться. Несомненно, что важную роль в выявлении больных с САТ играет знание врачом фенотипических признаков заболевания, что в сочетании с клиникой и специфическими изменениями на ЭКГ позволит быстро найти путь к правильному диагнозу и лечению.
Довольно трудной задачей является посмертный диагноз у больных, умерших от врожденных сердечных аритмий. По нашему мнению, характерные фенотипические признаки САТ могут быть включены в ряд симптомов при описании внезапно умерших молодых лиц, могут помочь в выявлении родственников с САТ и своевременном предотвращении у них угрожающих жизни состояний и ВС.



{kind=link}
{kind=link}
{kind=link}
{kind=link}