Амилоидоз – наследственное или приобретенное заболевание, обусловленное внеклеточным отложением в различных органах и тканях белково-полисахаридного комплекса с фибриллярной структурой – амилоида. Обладая химической инертностью и иммунологической толерантностью, амилоид накапливается в тканях, что приводит к атрофии и склерозу органов с развитием их функциональной недостаточности [1, 2, 3].
Распространенность амилоидоза до настоящего время не изучена ввиду не диагностированных случаев заболевания. В мире зафиксировано около 8 млн больных амилоидозом, что составляет 1% в популяции.
В основу современной классификации амилоидоза (ВОЗ, 2003) положен принцип специфичности основного фибриллярного белка амилоида [4]. Согласно данной классификации, вначале указывается тип амилоида, затем известный белок-предшественник и клинические формы амилоидоза с вовлеченными органами-мишенями. В настоящее время известно около 30 таких белков [5, 6, 7].
Поражение сердца может наблюдаться при AL – амилоидозе легких цепей, SSA – сенильном системном амилоидозе, FAP – семейном амилоидозе, АА – вторичном амилоидозе. FAP – наследственные системные амилоидозы – объединяют генетически наследуемые аутосомно-доминантные заболевания, при которых амилоидные фибриллы образованы мутантными формами транстиретина (ATTR – транстиретиновый амилоидоз), аполипопротеина A1 (AApoAI), аполипопротеина A2 (AApoAII), лизоцима (ALys), цистатина C (ACys), альфа-цепями фибриногена (AFIB) и т.д.
Семейный амилоидоз в большинстве случаев манифестирует к середине жизни, однако быстрое прогрессирование клинических проявлений в течение 5–15 лет приводят к развитию нейропатии, почечной, печеночной и сердечной недостаточности [3, 5]. Отложение амилоида в миокарде, эндокарде, перикарде, аорте и стенках коронарных сосудов приводит к развитию амилоидной кардиомиопатии с тяжелой сердечной недостаточностью [3, 4]. Среди FAP-форм амилоидоза более широко представлен ATTR-амилоидоз (транстиретиновый) – неуклонно прогрессирующее заболевание с неблагоприятным прогнозом [1, 3].
Распределение амилоида и характер поражения органов и систем зависит от типа мутации. Различают три основных фенотипа ATTR- амилоидоза:
- семейную амилоидную (транстиретиновую) полинейропатию;
- семейную амилоидную (транстиретиновую) кардиомиопатию;
- семейный лептоменингеальный (транстиретиновый) амилоидоз. Амилоидная трастиретиновая кардиомиопатия при наследственном ATTR-амилоидозе сопровождается выраженной сердечной недостаточностью [8]. Низкая осведомленность врачей по поводу этой орфанной патологии приводит к поздней диагностике ATTR-амилоидоза, когда уже развиваются необратимые изменения и тяжелая дисфункция органов. Ниже представлен клинический случай хронической сердечной недостаточности на фоне транстиретиновой амилоидной кардиомиопатии.
ОПИСАНИЕ КЛИНИЧЕСКОГО СЛУЧАЯ
Пациент Б., 51 года, впервые госпитализирован с жалобами на быструю утомляемость и одышку при малейшей физической активности, отеки лодыжек и стоп, онемение I, II, III пальцев рук, потерю массы тела до 6 кг в последние 6 мес. Обращало на себя внимание отсутствие предшествующего анамнеза кардиальных заболеваний, в том числе артериальной гипертензии. Клиническая симптоматика манифестировала в течение месяца.
Семейный анамнез по сердечно-сосудистой патологии не отягощен.
При объективном осмотре выявлено нормостеническое телосложение пациента, индекс массы тела (ИМТ) – 24 кг/м2.
Общее состояние средней степени тяжести, кожные покровы обычной окраски и влажности. В легких в нижних отделах справа отмечалось укорочение перкуторного звука и ослабление дыхания. Частота дыхательных движений (ЧДД) – 19/мин.
Границы сердца расширены, наблюдалось смещение верхушечного толчка влево. Тоны сердца приглушены, выявлялся систолический шум на верхушке с иррадиацией в левую подмышечную область. Частота сердечных сокращений (ЧСС) – 70/мин., пульс ритмичный. Артериальное давление (АД) – 123/71 мм рт ст.
Живот мягкий, при пальпации умеренная болезненность в правом подреберье. Печень выступает из-под края реберной дуги на 4 см. Селезенка не пальпируется. Стул регулярный. Мочеиспускание свободное, безболезненное. Отмечались отеки нижней трети голеней и стоп.
Данные ЭКГ: ритм синусовый, правильный. ЧСС – 72/мин. Низкий вольтаж QRS в стандартных отведениях. Электрическая ось сердца (ЭОС) отклонена влево. PQ – 0,22 c; QRS=0,10 с. Зубец Т отрицательный в I, II, aVL. Обнаружена блокада левой ножки пучка Гиса (рис. 1).
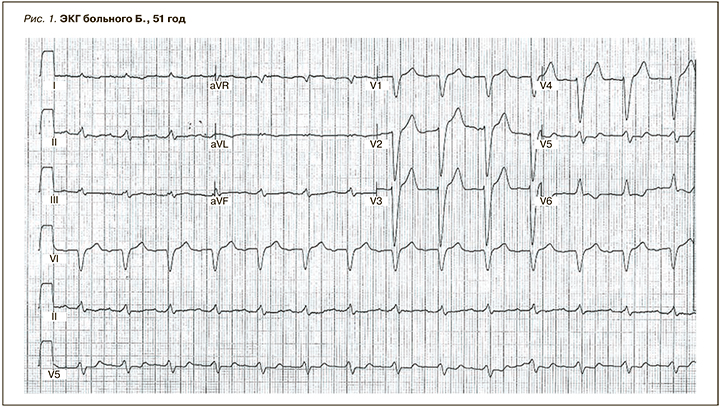
При рентгенографии органов грудной клетки выявлены признаки застойных явлений в малом круге кровообращения. Тень сердца расширена в обе стороны. Кардиоторакальный индекс (КТИ) – 76%.
По результатам лабораторного исследования выявлено небольшое повышение скорости оседания эритроцитов (СОЭ) до 25 мм/ч, аспартатаминотрансферазы (АСТ) – до 52,4 Ед./л, общего билирубина – до 29,9 мкмоль/л, щелочной фосфатазы – до 102,0 Ед/л, а также незначительное снижение уровня общего белка до 58,2 г/л.
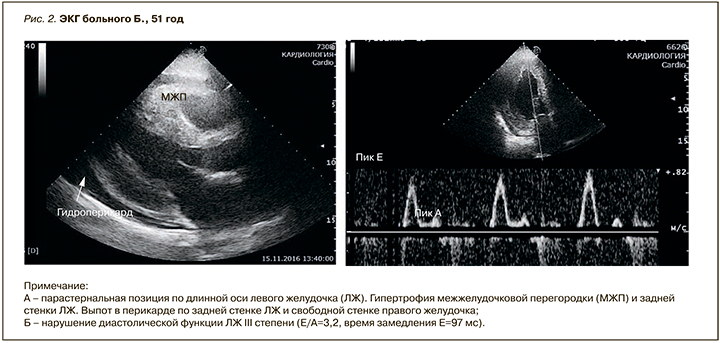
По данным эхокардиографии (ЭхоКГ) выявлена выраженная концентрическая гипертрофия левого желудочка (ЛЖ) с толщиной межжелудочковой перегородки (МЖП) до 1,8 мм и задней стенки до 1,7 мм, индексированная масса миокарда ЛЖ – 164 г/м2. Индекс относительной толщины стенки ЛЖ – 0,65. Конечно-диастолический объем ЛЖ составил 90 мл, конечно-систолический – 64 мл, фракция выброса (ФВ) – 29%. Обнаружены гипокинез всех сегментов ЛЖ, увеличение обоих предсердий. Конечно-систолический объем левого предсердия – 63 мл/ м2, правого – 42 мл/м2. Систолическое давление в легочной артерии – 43 мм рт.ст. Выявлены небольшая недостаточность митрального и трикуспидального клапанов, диастолическая дисфункция ЛЖ III степени (Е/ А=3,2, время замедления Е=97 мс). В полости перикарда умеренное количество выпота (рис. 2).
Результат УЗИ плевральных полостей: двусторонний гидроторакс.
УЗИ брюшной полости: гепатоспленомегалия. Нефролитиаз почек.
Таким образом, жалобы, данные объективного и инструментального обследования свидетельствовали о том, что у больного Б., 51 года, имеет место ХСН с низкой ФВ (ФВ=29%). Данные ЭхоКГ указывали на значительную концентрическую гипертрофию ЛЖ с нарушением диастолической функции III степени при отсутствии артериальной гипертензии или любого другого кардиологического заболевания в анамнезе. С учетом выявленных изменений по данным ЭхоКГ необходимо было исключить генетические аномалии сердца, в первую очередь рестриктивную кардиомиопатию и инфильтративные заболевания сердца. В таких ситуациях для уточнения характера поражения сердца рекомендуется МРТ.
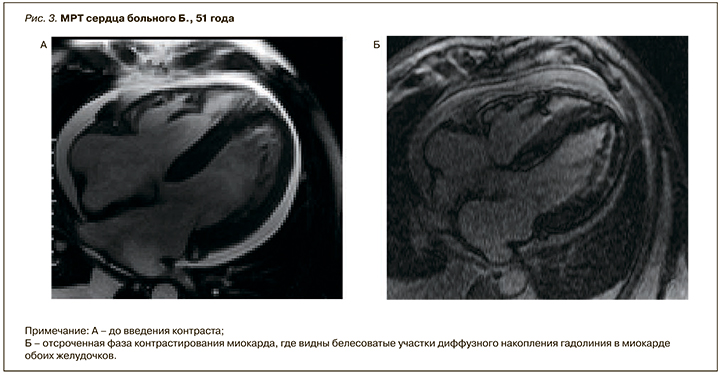
Данные МРТ сердца с гадолинием: отмечается симметричная гипертрофия миокарда ЛЖ, толщина МЖП в средней трети – 19 мм, толщина миокарда верхушки – 7 мм, толщина задней стенки ЛЖ – 15 мм. Сократимость миокарда ЛЖ значительно снижена за счет гипокинеза всех стенок (ФВ – 15%). Признаков обструкции полости ЛЖ, выносящего тракта ЛЖ нет. В верхней трети МЖП определялись клефты шириной основания 5 мм, глубиной до 9 мм. После внутривенного введения контрастного препарата в отсроченную фазу (через 10 мин) было обнаружено диффузное контрастирование миокарда ЛЖ, межпредсердной перегородки и правого желудочка, более выраженное по переднебоковой стенке, в МЖП со стороны правого желудочка. Описанная картина наиболее характерна для амилоидоза сердца (рис. 3).
В связи с тем что поражение сердца наиболее часто наблюдается при AL- и транстретиновом амилоидозе, пациенту для исключения моноклональной гамапатии были проведены электрофорез белковых фракции, пункция костного мозга и иммунохимическое исследование белков сыворотки и мочи. По результатам этих исследований моноклональной секреции выявлено не было.
Данные биопсии ткани слюнной железы: окраска конго-красным с выявлением депозитов амилоида реакция положительная.
Результаты генетического теста на семейную амилоидную кардиомиопатию: методом прямого секвенирования проведено исследование всей кодирующей последовательности и областей экзон-интронных соединений гена TTR, ответственного за развитие наследственного амилоидоза. В результате анализа в экзоне 2 гена TTR выявлен патогенный вариант c.128G >A в гетерозиготном состоянии.
При каскадном семейном генетическом скрининге у одного из троих детей пациента (мальчик 11 лет) также обнаружена мутация гена S23N в экзоне 2, ответственная за транстиретиновый амилоидоз.
Таким образом, на основании молекулярно-генетического обследования пациенту был поставлен клинический диагноз:
- основной: наследственный транстиретиновый амилоидоз с мутацией в S23Nв экзоне 2. Амилоидная кардиомиопатия. Периферическая амилоидная полинейропатия 1 стадии;
- осложнения: ХСН IIБ ст., ФК III NYHA. Блокада левой ножки пучка Гиса. Двусторонний гидроторакс. Гидроперикард. Асцит.
Пациенту были назначено следующее лечение:
- Специфическая терапия ATTR-амилоидоза – тафамидис, который ограничивает диссоциацию нативного ТTR тетрамера в мономеры и ингибирует образование амилоидных фибрилл транстиретина. Впоследствии под действием этого лекарственного средства прекращается дальнейший синтез амилоида и его накопление [10–11].
- Симптоматическая терапия петлевыми диуретиками (торасемид 20 мг/сут), антагонистами минералокортикоидных рецепторов (спиронолактон 50 мг/ сут). Ингибиторы АПФ не использовались в связи с низкими значениями АД и их неблагоприятным влиянием на гемодинамику больных амилоидной кардиомиопатией, провокацией ортостатических реакций. Бета-адреноблокаторы не применялись по причине низкого АД у пациента и склонности к брадикардии.
В дальнейшем пациент неоднократно госпитализировался с явлениями застойной сердечной недостаточности. Течение ХСН осложнилось развитием синусовой брадикардии с эпизодами головокружения. По данным суточного мониторирования ЭКГ (СМЭКГ) наблюдались суточные колебания ЧСС от 33 до 58 в мин; средняя ЧСС составила 44 уд./мин. Выявлены эпизоды неустойчивой желудочковой тахикардии, фибрилляции предсердий и АВ-блокада I–II степени (рис. 4).

Пациенту установлен электрокардиостимулятор с частотной адаптацией (ЭКС DDDR) с функцией кардиовертера-дефибриллятора (ИКД) (рис. 5).
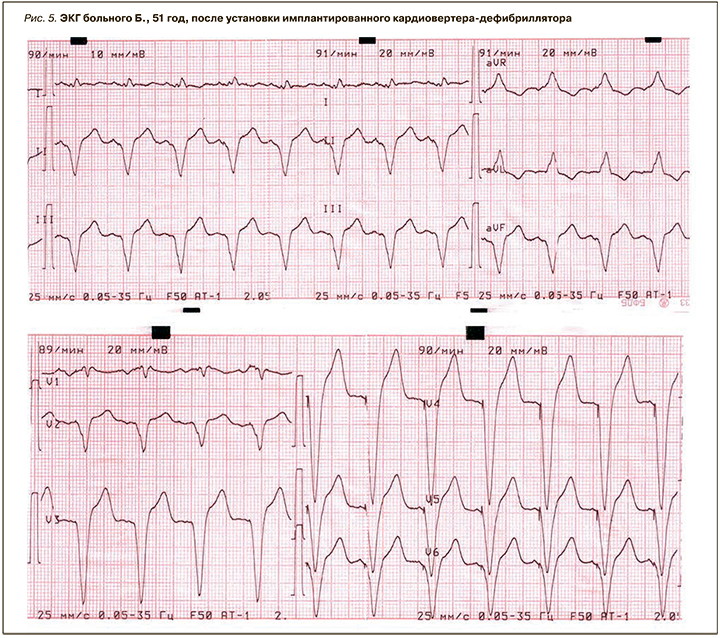
После установки ИКД состояние пациента существенно улучшилось: ЧСС увеличилась до 60–84 в мин, повысилась толерантность к физической нагрузке, головокружение не беспокоило.
ОБСУЖДЕНИЕ
АТТR-вариант семейного амилоидоза – редкое заболевание, которое ранее встречалось только у афроамериканцев [6]. В последнее время единичные случаи этой патологии регистрируются и в России. Несмотря на наследственную природу АТТR-вариант наследственного амилоидоза проявляется только в середине жизни [5, 7], что и наблюдалось в приведенном клиническом примере. Этот клинический случай демонстрирует течение семейной амилоидной (транстиретиновой) кардиомиопатии, подтвержденная молекулярно-генетическими методами.
По данным литературы, у больных с наследственным ATTR-амилоидозом чаще всего выявляют поражение периферической (синдром туннельного канала, сенсомоторная периферическая полинейропатия, миалгии, потеря мышечной массы) и вегетативной (ортостатическая гипотония с развитием обморочных состояний, моторная диарея, нарушения потоотделения и др.) нервной системы [9]. В данном клиническом случае поражение нервной системы представлено незначительно выраженными симптомами периферической нейропатии в виде дизестезии I, II, III пальцев рук и потери массы тела. Склонность к артериальной гипотонии, вероятно, обусловлено автономной дисфункцией.
Известно, что амилоидоз вызывает нарушения проводимости сердца начиная от бессимптомной атриовентрикулярной блокады и ножек пучка Гиса до тяжелой и быстро прогрессирующей сердечной недостаточности. Это обусловлено способностью амилоида проникать в любую структуру сердца, включая проводящую систему, миокард желудочков и предсердий, а также в клапанную ткань с формированием недостаточности клапанов. На ранних стадиях заболевание может протекать бессимптомно, проявляясь лишь утолщением стенки ЛЖ при ЭхоКГ. При прогрессировании амилоидоза симптоматика быстро нарастает, особенно после интеркуррентного заболевания [9]. В нашем клиническом примере поражение амилоидом проводящих путей сердца подтвердилось данными ЭКГ и СМЭКГ (синусовая брадикардия с минимальной ЧСС 33/мин, атриовентрикулярная блокада I–II степени).
Лечение амилоидоза представляет собой трудную задачу. При наследственном ATTR-амилоидозе трансплантация печени – органа, где синтезируется практически весь транстиретин организма, – это удаление источника мутантного транстиретина. Этот метод лечения длительное время считался единственной эффективной мерой при ATTR-амилоидозе [10, 11]. Однако выяснение механизмов, способствующих формированию амилоидных депозитов и неправильной свертываемости белков, а также механизмов ТТR-тетрамерной стабилизации, послужило основанием для разработки ТТR-стабилизирующих агентов. Таким модифицирующим агентом является тафамидикс – кинетический стабилизатор транстерина. Он ограничивает диссоциацию нативного ТTR тетрамера в мономеры и ингибирует образование амилоидных фибрилл TTR.
ЗАКЛЮЧЕНИЕ
Таким образом, в рассмотренном клиническом случае у пациента наблюдался АТТR-вариант семейного амилоидоза с преобладанием кардиальных симптомов и развитием сердечной недостаточности. Продемонстрирован случай диагностики и верификации редкой патологии – АТТR-амилоидоза сердца с помощью современных методов исследования. ATTR-амилоидоз имеет тяжелое и прогрессирующее течение и без адекватного и своевременного лечения способствует развитию осложнений, которые могут привести к летальному исходу. Своевременное выявление амилоидоза и раннее начало лечения для замедления прогрессирования заболевания очень важно, так как это определяет продолжительность и качество жизни, прогноз заболевания и дальнейшую тактику ведения.
