Моноклональные антитела (МАТ) представляют собой один из способов таргетного воздействия на патогенетические механизмы тех или иных заболеваний. Наибольшее распространение лечение препаратами этой группы получило в онкологии и онкогематологии, однако в последнее десятилетие схемы лечения с применением антител входят также в клинические рекомендации по лечению различных внутренних болезней. В первую очередь это ревматология, где, например, используются антитела к рецептору интерлейкина-6 (IL-6) тоцилизумаб и сарилумаб. Другие области применения этой группы лекарственных средств – бронхиальная астма (омализумаб – антитела к IgE), воспалительные заболевания кишечника (ведолизумаб – антитела к α4β7-интегрину); остеопороз (деносумаб – антитела к лиганду рецептора RANK).
В кардиологии известное распространение получили снижающие уровень липопротеидов низкой плотности (ЛПНП) антитела к пропротеин конвертазе субтилизин/кексин типа 9 (PCSK-9) алирокумаб и эволокумаб, абцисимаб (антиагрегант, связывающий GPIIb/IIIa рецепторы тромбоцитов), Fab-фрагменты человеческих антител, связывающие дигоксин и дабигатран (идаруцизумаб).
Клинический эффект МАТ во многом зависит не только от их структуры, определяющей аффинитет к тем или иным мишеням, но и особенностей фармакокинетики.
Антитела представляют собой гетеродимерные протеины, состоящие из двух тяжелых цепей (50 кДа для изотипа G) и двух легких цепей (25 кДа), связанных остатками цистина. Связанные цепи формируют γ-образную молекулу. Каждая цепь молекулы антитела имеет домены вариабельной структуры (VH и VL для тяжелой и легкой цепей), а также домены постоянной структуры (CL, CH1, CH2 и CH3). Два фрагмента антитела, образованные каждый двумя вариабельными и двумя постоянными доменами тяжелой и легкой цепей, называют Fab-фрагментами, или антиген-связывающими фрагментами. Непосредственно интерфейс, сформированный вариабельными доменами на конце Fab-фрагмента, называется паратоп – сайт распознавания антигена антителом. В свою очередь участок с постоянной структурой, состоящей из CH2 и CH3-доменов двух тяжелых цепей, формирует так называемый кристаллизующийся фрагмент (Fc – fragment crystallizable), который может связываться как с компонентами комплемента (например, C1q), так и различными клеточными рецепторами, включая (но не ограничиваясь) неонатальный рецептор (FcRn) и рецептор FcγRs [1].
Химерные антитела (цетуксимаб и ритуксимаб) содержат VL и VH из молекул, полученных от мышей, и СL1–3 и CH1–3, имеющих человеческое происхождение. Дальнейшее замещение структурных компонентов, полученных от мышей, на их человеческие аналоги привело к появлению гуманизированных антител (например, трастузумаб и алемтузумаб).
В итоге появление полностью человеческих антител стало возможным благодаря использованию методики фагового дисплея для отбора структур вариабельных доменов [2].
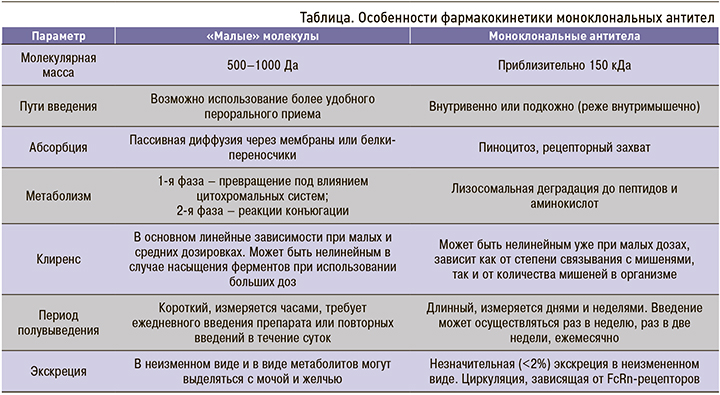
В отличие от малых молекул, фармакокинетика МАТ имеет свои существенные отличия, которые обусловлены сочетанием различных факторов, определяющих распределение и пути элиминации этих молекул (табл.).
ВВЕДЕНИЕ МОНОКЛОНАЛЬНЫХ АНТИТЕЛ
МАТ не могут применяться перорально ввиду следующих обстоятельств:
- крупные молекулы крайне плохо (не более 1–2%) всасываются в желудочно-кишечном тракте и не могут создавать предсказуемой системной концентрации;
- белковые структуры подвергаются денатурации в кислой среде желудка;
- в результате денатруации сложные белковые структуры теряют защиту от протеаз [3, 4].
Основные пути введения МАТ в организм – внутривенные и подкожные инъекции.
Всасывание и распределение при введении препаратов под кожу зависят от их способности проникать из интерстициального пространства в лимфатические сосуды. Из-за того что ток лимфы значительно медленнее тока крови, а также с учетом пассивности процесса проникновения крупных молекул в лимфатические сосуды время регистрации максимальной концентрации препарата в крови после его подкожного введения может быть отсрочено и составляет от 2 до 14 дней (при этом для большинства МАТ этот показатель составляет 6–8 дней) [5].
Необходимо также отметить, что при подкожном введении МАТ могут подвергаться пресистемной элиминации за счет присутствия в интерстициальной жидкости протеаз, эндоцитоза и последующей лизосомальной деградации в клетках эндотелия лимфатических сосудов. Кроме того, одним из ведущих путей пресистемной элиминации при этом пути введения является фагоцитоз в лимфатических узлах. Все это определяет уменьшение биодоступности МАТ, которое, по разным данным, составляет от 50 до 80% [5, 6].
На биодоступность МАТ влияет выбор места для инъекций, разная скорость лимфотока, изменяющаяся, например, при активных движениях, различное состояние лимфатических узлов. Нужно также учитывать влияние возраста, пола, состояния кожи, уровня артериального давления и частоты сердечных сокращений, частоты дыхания. Все эти факторы, которые учитываются при фармакокинетическом моделировании, основанном на физиологических особенностях, уменьшают предсказуемость пресистемных потерь при подкожном введении и влияют на разброс концентрации препарата в системном кровотоке [6].
Этих недостатков лишено внутривенное введение МАТ, при котором препарат поступает сразу в центральную камеру. Однако последнее может быть связано как с известными неудобствами для пациентов, так и непредсказуемыми реакциями, обусловленными достижением пиковых концентраций. В качестве примера может рассматриваться «цитокиновый шторм», развившийся у здоровых добровольцев в ходе проведения исследований препарата TGN1412 [7]. Нужно заметить, что изменение дозы и скорости введения препарата в дальнейшем позволило вернуться к его применению у человека и дальнейшему изучению [8]. Поэтому при внутривенном введении МАТ ключевыми параметрами безопасности могут быть не только однократная доза, но и скорость ее введения.
РАСПРЕДЕЛЕНИЕ МОНОКЛОНАЛЬНЫХ АНТИТЕЛ
Процесс распределения МАТ в организме зависит от нескольких факторов, а именно их способности проникать сквозь стенки сосудов в интерстициальные ткани, связываться на поверхности клеток с рецепторами и проникать внутрь клетки.
В связи с большим размером молекул и их зарядом процесс необлегченного проникновения МАТ сквозь клеточные мембраны затруднен и имеет незначительный вклад в фазу распределения в организме. Молекулы с массой более 16 кДа имеют крайне ограниченные возможности для прямого проникновения через сосудистую стенку [9].
Главным механизмом, обеспечивающим выход этих молекул из сосудистого русла, служит водная конвекция, осуществляющаяся под влиянием различий в уровнях гидростатического давления при условии наличия пор в межклеточных соединениях. Кроме того, на процесс перехода МАТ в интерстициальное пространство и лимфу влияет разница в онкотическом давлении и заряд молекул: чем большим отрицательным зарядом обладает макромолекула, тем сложнее ей покинуть сосудистое русло [10].
Другим важным процессом, влияющим на распределение, является рецептор-опосредованный трансцитоз. Главным образом этот процесс осуществляется путем подключения неонатального рецептора (FcRn). При этом необходимо принимать во внимание, что рецептор-опосредованный трансцитоз может осуществляться в обоих направлениях – как апикобазальном (из просвета сосуда), так и в базальноапикальном (в просвет сосуда) [11, 12].
Возможности проникновения МАТ во внеклеточный матрикс ограничиваются его характеристиками. Последний представляет собой сеть, формирующуюся преимущественно отрицательно заряженными глюкозаминогликанами (например, гиалуроновой кислотой), присутствие которых ограничивает проникновение и накопление крупных, а также отрицательно заряженных белков. Массовое соотношение глюкозаминогликанов, формирующих матрикс, и белковых молекул, проникающих из сосудистого русла, обычно составляет 1:1 (максимальный исключающий объем для IgG составляет 50%) [13].
Следующий фактор, который может влиять на распределение МАТ, – степень связывания с мишенями. Здесь необходимо учитывать, что таргетные молекулы или рецепторы могут располагаться как вне сосудистого русла (чаще), так и внутри него (например, цитокины или циркулирующие компоненты комплемента).
Подчеркнем, что объем кажущегося распределения (Vd ‒ volume of distribution) зависит от связывающей способности рецепторов. При ограниченном уровне этого показателя с увеличением плазменной концентрации вводимого препарата связь концентрации с объемом распределения перестает быть линейной, т.е. Vd уменьшается за счет снижения связывающей способности рецепторов и увеличения в кровеносном русле содержания свободного препарата.
Дальнейшим этапом распределения антител является их проникновение из интерстициальных тканей в лимфу. Так же, как и в случае с выходом из кровеносного русла, этот процесс зависит от градиентов давлений, скорости лимфотока и эффекта проникновения (sieving effect) [14].
Объем кажущегося распределения МАТ во многом зависит от распределения растворимых и поверхностных вариантов мишеней. Так, у препаратов, для которых мишени сосредоточены в большей степени в интерстициальном пространстве, объем распределения может составлять 6–30 л, что говорит о распределении препарата между двумя камерами в соотношении от 1:1 до 8:1. При оценке объемов центральной и периферической камер (для двухкамерной модели) объем первой составляет 2–3 л (что соответствует объему плазмы), а объем периферической камеры в стационарном состоянии колеблется от 8 до 20 л [6, 15, 16].
У препаратов, которые имеют высокую аффинность к определенным тканям (например, головному мозгу), этот показатель может составить до 300 л (примерно 1:100) [17].
Для моделирования процессов распределения может быть введен показатель проникновения молекул через капилляры в зависимости от плотности контактов между эндотелиоцитами, который оценивается как 1-σ, где σ – показатель, пропорциональной плотности межклеточных контактов. Для мышц, кожи, соединительной ткани он составляет 0,95, в то время как для тканей с сетчатой структурой эндотелия (печень, селезенка, костный мозг) от 0,31 до 0,42 [18].
ЭЛИМИНАЦИЯ МОНОКЛОНАЛЬНЫХ АНТИТЕЛ
В отличие от малых молекул, которые могут экскретироваться в неизменном виде, в основе элиминации МАТ лежит их катаболизм. Проникновение крупных белковых молекул через боуменову капсулу почек и их экскреция в неизмененном виде в норме практически отсутствует. Это связано с тем, что мембрана почечного клубочка не пропускает молекулы массой более 55 кДа. Фильтроваться могут относительно мелкие фрагменты антител, но они реабсорбируются в дистальных участках петли нефрона, где подвергаются дальнейшим метаболическим превращениям [19].
С желчью может экскретироваться также только незначительная часть IgG [20].
Таким образом, основным путем элиминации антител служит внутриклеточная элиминация, реализующаяся последствием деградации молекул до составляющих их аминокислот в лизосомах. Проникновение антител внутрь клетки происходит как путем неспецифичного пиноцитоза, так и пиноцитоза, который является рецептор-опосредованным и отмечается после связывания антитела со структурами клеточной мембраны. Связывание это может осуществляться как Fc-, так и Fab-фрагментами [21].
Неспецифический пиноцитоз – один из главных путей проникновения антител внутрь клетки. Большая поверхность эндотелия в организме человека (>1000 м2/70 кг) приводит к экстенсификации этого процесса путем многократного прохождения растворенных антител через богатые эндотелием ткани. Таким образом, значительное количество антител элиминируется мышцами, кожей и тканями желудочно-кишечного тракта даже в отсутствие специфического аффинитета к клеткам этих тканей и особенностей распределения [22].
Если связывание осуществляется через комплемент-детерминирующий участок Fab-фрагмента антитела и реализуется посредством присоединения к специфическому для антитела эпитопу (мишени), такой механизм носит названия мишень-опосредованной диспозиции (TMDD – target mediated drug disposition). В этом случае скорость элиминации зависит от нескольких аспектов:
- экспрессии таргетных рецепторов на поверхности клетки;
- степени аффинности антитела конкретным рецепторам;
- концентрации антител;
- способности клеток к пиноцитозу;
- активности лизосомальных ферментов клетки [21].
Для большинства МАТ, имеющих терапевтическую мишень, локализованную на поверхности клетки, подобный путь является основным путем элиминации при введении в малых и средних терапевтических дозах, когда зависимость скорости элиминации от концентрации препарата имеет линейный характер. При увеличении дозировок развивается насыщение рецепторов, после чего эта связь перестает быть линейной (ментеновская кинетика или фармакокинетика с насыщением), и элиминация «избыточного» количества антител происходит альтернативными путями.
В частности, одним из возможных путей выступает элиминация, осуществляемая клетками иммунной системы (моноцитами, макрофагами, дендритными клетками) вследствие связывания Fc-фрагмента антитела с локализованными на мембране этих клеток рецепторами FcγRs.
В этом случае процесс рецептор-опосредованного пиноцитоза с дальнейшим лизосомальным катаболизмом является сходным с процессом мишеньопосредованной диспозиции [23].
Следует отметить, что FcγRs-опосредованный путь элиминации не вносит существенный вклад в фармакокинетику антител, которые не образуют растворимые иммунные комплексы, в то время как в случае образования последних (например, элотузумаб) этот путь элиминации может быть основным [24].
С учетом того что элиминация антител может быть во многом неспецифичным процессом, существенную роль могут играть механизмы, защищающие антитела от быстрой деградации и повышающие период их циркуляции в системном кровотоке. Одним из таких механизмов является связь с так называемым Брамбелл-рецептором (рецептором FcRn) [25]. Если pH среды физиологическая, связь IgG с этим рецептором не является существенной, однако же аффинитет повышается при уменьшении pH, что характерно для кислой среды рибосомы. В этом случае образующийся комплекс FcRn–IgG выводится из лизосомы и из клетки обратно во внеклеточную среду, что обес-печивает возврат из периферического в центральный компартмент и удлиняет время циркуляции антител. Подобный механизм защиты от деградации в лизосомах характерен более чем для двух третей использующихся в настоящее время моноклональных антител [26].
Так, период полувыведения (T1/2) антител, относящихся к классам IgG1, IgG3 и IgG4, может составлять 2–3 нед, что в разы превышает аналогичный показатель для других белков со сходной молекулярной массой. В то же время T1/2 антител класса IgG2 составляет не более 7 дней, что связано с отсутствием реализующегося через Брамбелл-рецептор защитного механизма [27].
Насыщение FcRn с уменьшением протективного эффекта достигается в эксперименте при существенном превышении концентрации вводимых иммуноглобулинов выше физиологических показателей. Такое состояние может отмечаться, например, при наличии у пациентов множественной миеломы. Однако для большинства МАТ доза не превышает 10 мг/кг, что составляет не более 2% от физиологического содержания IgG в организме. Таким образом реализуемый FcRn протективный эффект сохраняется [28].
ОСОБЕННОСТИ ФАРМАКОДИНАМИКИ, СВЯЗАННЫЕ С МИКРОГЕТЕРОГЕННОСТЬЮ
Одна из типичных причин различий внутри идентичных белковых молекул (под идентичностью подразумевается одинаковая аминокислотная последовательность, также и сходные третичная и четвертичная структуры) – посттрансляционные модификации. Такие модификации могут определять стабильность белка в клетке, регулировать биологическую активность, влиять на взаимодействия с другими биологическими молекулами, опосредовать иммуногенность. С одной стороны, эти модификации могут быть необходимым условием созревания белка в качестве биологически активной единицы, а с другой ‒ они могут приводить к его инактивации [29].
Присоединение к белковой молекуле тех или иных функциональных групп может влиять на ее заряд, что приводит к изменению фармакокинетических свойств. Так, анионные модификации (присоединение групп с отрицательным зарядом) уменьшают плазменный клиренс и повышают период пребывания препарата в центральной камере, в первую очередь за счет уменьшения неспецифического пиноцитоза [30]. В то же время катионные модификации повышают плазменный клиренс и ускоряют фазу тканевого распределения препарата. При этом белки, подвергнутые в большей степени катионным модификациям, хуже всасываются при подкожном введении [31].
Существенным влиянием на фармакокинетические параметры обладают паттерны гликозилирования антител. В зависимости от них может меняться как аффинность к рецепторам и соответственно рецептор-опосредованный клиренс, так и степень защищенности антитела от воздействия протеаз.
Механизмы посттрансляционной модификации лежат в основе микрогетерогенности получаемых продуктов. Этот термин обозначает ситуацию, когда в любом образце биологического препарата, полученного из одного источника и максимально очищенного от родственных примесей, некоторые структурные параметры варьируют в определенных пределах, т.е. препарат представляет собой смесь целевого белка и родственных веществ [29].
Например, для трастузумаба можно выделить восемь различных изоформ [32]. Степень микрогетерогенности, присущая препаратам даже внутри одной промышленной серии, может увеличиваться при сравнении между собой различных серий и предположительно оказывается еще выше при выпуске продукта на разных производственных площадках. Необходимо принимать во внимание, что микрогетерогенность потенциально способна не только изменять фармакокинетические и фармакодинамические свойства препарата, но и влиять и на иммунный ответ организма.
ИЗМЕНЕНИЯ ФАРМАКОКИНЕТИКИ У ПАЦИЕНТОВ В ПЕРИОД ЛЕЧЕНИЯ
Одним из основных механизмов, потенциально влияющих на фармакокинетику МАТ, может быть иммунная реакция, в результате которой нарастает клиренс, опосредованный иммунной системой организма [6].
Интересно, что эта же реакция используется в процессе направленной эволюции белков, когда с целью улучшения свойств имеющихся молекул получают новые антитела с улучшенными связывающими свойствами, например, связывающимися с компонентами комплемента. Общее влияние на фармакокинетику здесь таково: чем больше вероятность связывания антитела, тем выше ее плазменный клиренс. Это объясняет то, почему некоторые новые антитела имеют T1/2 меньше характерного в целом для этой группы периода, равного 2–3 нед [33].
В целом же особенности клиренса МАТ таковы, что скорость их выведения во многом зависит от степени связи с рецепторами, в том числе теми, которые являются основной мишенью препарата и через которые реализуется его фармакодинамический эффект. Закономерно, что чем больше количество экспонированных на клеточной поверхности мишеней или в случае онкологической патологии чем больше клеточная масса опухоли, тем большее количество препарата распределится из плазмы в ткани. При этом в ходе терапии у пациентов, ответивших на лечение, отмечается феномен уменьшения плазменного клиренса, связанный с уменьшением массы мишеней/клеток носителей мишеней. То же отмечается при применении антител, направленных на снижение системного воспалительного ответа. Косвенным признаком отсутствия терапевтического ответа на проводимое лечение в этих случаях служит отсутствие уменьшения плазменного клиренса с течением времени ‒ это может говорить о сохраняющемся количестве мишеней [34].
ФАРМАКОКИНЕТИЧЕСКОЕ МОДЕЛИРОВАНИЕ
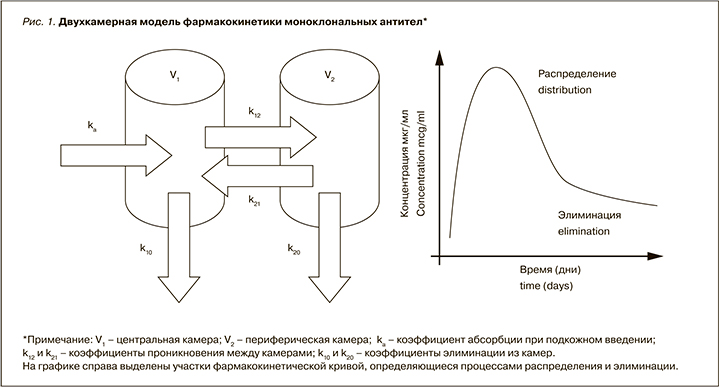
Наиболее простой моделью, описывающей фармакокинетику МАТ, служит двухкамерная модель с обратимым распределением препарата между центральной и периферической камерами (рис. 1). При этом коэффициенты проникновения k12 и k21 во многом зависят не только от структуры антитела, но и посттрансляционных модификаций и возможности реализации механизма выведения из лизосом комплекса IgG–FcRn, а коэффициент выведения из центральной камеры (k10) может быть значимо меньше аналогичного показателя для периферической камеры (k20) [6].
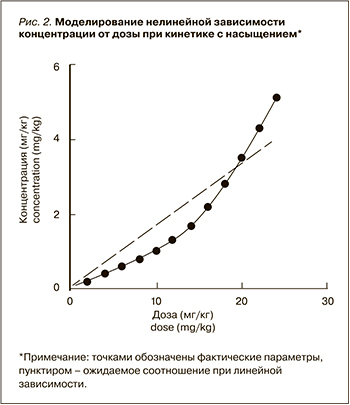
Процесс распределения из центральной камеры может быть описан кинетикой первого порядка для большинства дозовых режимов. В некоторых случаях, когда распределение определяется в первую очередь мишень-опосредованным механизмом, для описания может применяться уравнение Михаэлис–Ментен с определением максимальной скорости элиминации (Vmax) и константы элиминации km. На рисунке 2 приведены данные моделирования зависимости плазменной концентрации от вводимой дозы. При введении препарата в дозе выше 10 мг/кг зависимость перестает носить линейный характер, что связано с развитием насыщения мишеней и снижением скорости мишень-опросредованной элиминации из центральной камеры (уменьшение k12) [35].
 Примером достаточно сложных подходов являются попытки построения так называемых моделей, базирующихся на физиологических параметрах. Скажем, возможно выделение в качестве различных камер органов и тканей, сосудистый эндотелий в которых имеет либо более плотные, либо менее плотные межклеточные контакты. Плотность этих контактов определяет различия в степени свободного проникновения антител в интерстициальное пространство.
Примером достаточно сложных подходов являются попытки построения так называемых моделей, базирующихся на физиологических параметрах. Скажем, возможно выделение в качестве различных камер органов и тканей, сосудистый эндотелий в которых имеет либо более плотные, либо менее плотные межклеточные контакты. Плотность этих контактов определяет различия в степени свободного проникновения антител в интерстициальное пространство.
Кроме того, различия распределения препарата в камеры может быть обусловлено кровотоком и лимфотоком, рецепторной характеристикой и количеством иммунокомпетентных клеток (в последнем случае лимфатические узлы могут быть рассмотрены в виде отельной камеры).
В целом эти подходы требуют высокой степени детализации и мультипликации количества камер, когда в качестве отдельных камер выделяются не только сосуды и ткани, но даже клеточные субкомпартменты (например, лизосомы).
В качестве простой иллюстрации этого подхода можно с целью моделирования, базирующегося на физиологии, выделить четыре камеры: центральную (сосудистое русло) и три периферических (интерстициальное пространство, внутриклеточное пространство и эндосомы) [36].
ЗАКЛЮЧЕНИЕ
Фармакокинетика МАТ представляет собой отдельную область изучения свойств этих препаратов. В целом антитела характеризуются особенностями распределения в тканях организма, которые зависят не только от размера молекулы и аминокислотной последовательности, но и от аффинитета к рецепторам и от определяющих клиренс посттрансляционных модификаций.
На фармакокинетические параметры может влиять продолжительность лечения – это связано как с изменением количества мишеней (изменения мишень-опосредованного клиренса), так и возможной иммунизацией организма.
Несмотря на то что в первом приближении классическая двухкамерная модель описывает распределение и элиминацию МАТ, усложнение модели путем построения моделей, базирующихся на физиологических особенностях, требует высокого уровня детализации и даже включения субклеточных структур в качестве отдельно рассматриваемых камер.
