ВВЕДЕНИЕ
Орфанными (редкими) заболеваниями принято считать заболевания с распространенностью не более 10 случаев на 100 тыс. человек. Однако надо понимать, что пациента с редким заболеванием могут встретить врачи любой специальности. Именно терапевтическая служба обеспечивает первичный контакт с большинством пациентов, поэтому достаточная осведомленность врачей-терапевтов и врачей общей врачебной практики в диагностике орфанных заболеваний представляется чрезвычайно важной [1].
На сегодняшний день известно более 7000 редких заболеваний. Для большинства из них не существует эффективного лечения, однако существуют методы, позволяющие улучшить качество и продолжительность жизни пациентов. Именно поэтому исключительно большую роль играет ранняя диагностика орфанной патологии.
Большинство редких заболеваний имеют тяжелое хроническое течение с инвалидизацией и нередко летальными исходами. Симптомы могут быть очевидны с рождения или проявляться в детском возрасте. В то же время более половины редких заболеваний дают о себе знать уже во взрослом возрасте. Немаловажно то, что такие болезни часто протекают под маской других, более распространенных заболеваний. Основная задача врача терапевта – заподозрить редкую болезнь и направить больного для уточнения диагноза к узким специалистам.
Редкие болезни условно можно разделить на две группы:
- наследственные. Могут как передаваться от родителей, так и возникать из-за первичной мутации генов. Они не всегда проявляются сразу после рождения ребенка, поэтому нельзя смешивать понятия «врожденный» и «наследственный»;
- ненаследственные. Обусловлены либо факторами внешней среды, либо сочетанием этих факторов с наследственной предрасположенностью. Внешние причины мутаций могут быть различными – воздействие облучений, сверхвысоких частот, ультрафиолетового излучения, химических и токсических веществ.
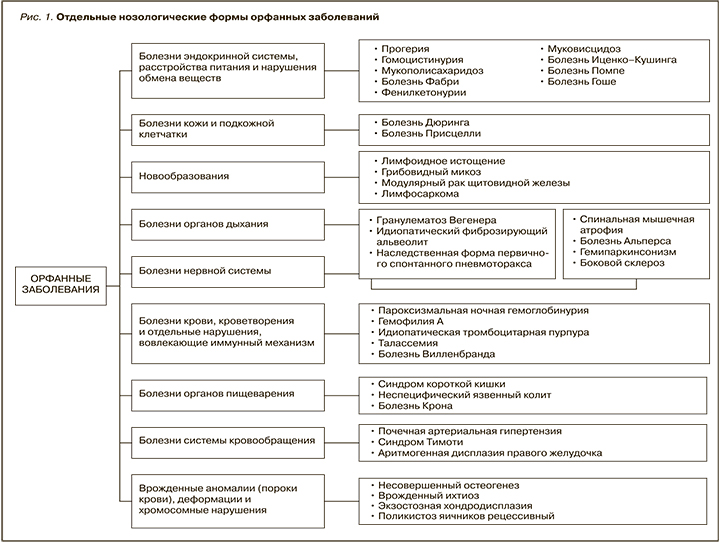
Рисунок 1 отражает спектр наиболее распространенных орфанных заболеваний и подчеркивает возможность вовлечения практически всех органов и систем в патологический процесс. Далее мы рассмотрим несколько относительно часто встречающихся орфанных заболеваний и диагностические алгоритмы их выявления.
БОЛЕЗНЬ ФАБРИ (Е75.2)
Болезнь Фабри, или болезнь Андерсона–Фабри, – наследственное заболевание, относящееся к группе лизосомных болезней накопления и обусловленное значительным снижением активности или отсутствием фермента альфа-галактозидазы [2]. Вследствие неправильного обмена веществ происходит поражение кожи, глаз, суставов, нервной системы, сердца, почек, желудочно-кишечного тракта (ЖКТ).
К ранним проявлениям болезни Фабри относятся:
- мелкие темно-красные мягкие узелки на коже, обычно в паху, внутри пупка, на слизистых;
- нейропатические боли;
- вихревидная кератопатия;
- слабое потоотделение или его отсутствие;
- желудочно-кишечные нарушения.
Полный спектр клинических проявлений этого заболевания представлен на рисунке 2.
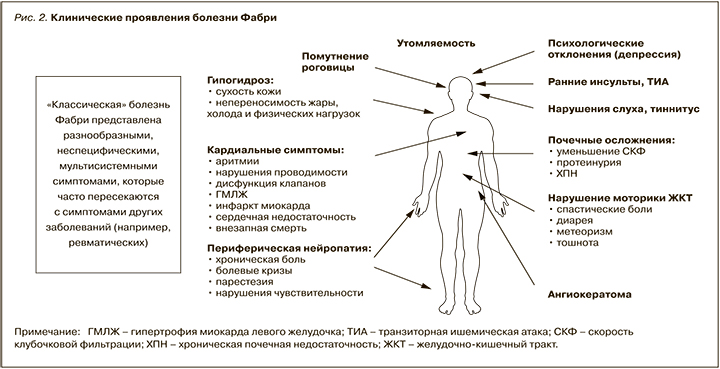
Первые симптомы заболевания возникают в детском и подростковом возрасте: у мужчин в 10 лет, у женщин – в 15. Однако следует помнить, что болезнь Фабри может проявиться и у взрослых: в возрасте около 20–40 лет у мужчин или даже в более позднем возрасте у женщин [3].
Первым типичным признаком заболевания является сыпь (ангиокератома) – яркие мелкие пятна, располагающиеся на коже передней брюшной стенки, спины, ягодиц, бедер, в области поясницы.
Второй типичный признак – акропарестезии, боли в кистях и стопах, которые появляются часто в жаркую погоду, при физической нагрузке, повышении температуры тела у человека. Пациенты эти ощущения сравнивают с прикосновением к раскаленной плите.
Третий типичный признак – снижение или отсутствие потоотделения, четвертый – желудочно-кишечные нарушения (понос, запор, боли в животе).
Параллельно с этим развивается поражение внутренних органов, однако клинически выраженным оно становится позднее. При болезни Фабри наиболее часто поражаются почки, сердце и головной мозг [3].
У кого еще можно заподозрить болезнь Фабри?
1. У пациентов с застойной сердечной недостаточностью, нарушением проводимости, стенокардией, гипертрофией миокарда (если причина утолщения непонятна).
2. У пациентов со сниженной функцией почек, вплоть до терминальной стадии почечной недостаточности.
3. У пациентов в возрасте до 50 лет, имеющих в анамнезе инсульт.
4. У пациентов с неврологической симптоматикой и с поражением других систем (сердца, почек, глаз).
5. У пациентов с неясной неврологической симптоматикой.
Помимо акропарестезий, у пациентов с болезнью Фабри могут наблюдаться боли в суставах и эпизоды необъяснимой лихорадки, сопровождающиеся лабораторными признаками воспаления. Повышение скорости оседания эритроцитов (СОЭ) и уровня С-реактивного белка у некоторых пациентов сохраняется практически постоянно, что позволяет обсуждать роль аутовоспаления в патогенезе заболевания. По мнению некоторых экспертов, болезнь Фабри следует включать в алгоритм обследования пациентов с лихорадкой неясного генеза [2]. Поражение кожи и нервной системы, особенно в сочетании с болями в суставах, лихорадкой, лабораторными признаками воспаления, нередко трактуется как проявление ревматических заболеваний.
Наибольшие трудности в диагностике представляет атипичный, или поздний, вариант болезни Фабри, когда у пациентов в возрасте 40–50 лет и старше выявляют поражение сердца при отсутствии классических симптомов этого заболевания, что обычно расценивается как гипертрофическая кардиомиопатия или вторичная гипертрофия левого желудочка, связанная с артериальной гипертонией. Типичные проявления болезни Фабри включают ишемический инсульт и транзиторные ишемические атаки, развивающиеся в молодом возрасте [3].
Все клинические признаки болезни Фабри неспецифичны, поэтому для подтверждения диагноза необходимы лабораторные и молекулярно-генетическое исследования. Для этого заболевания характерны снижение активности альфа-галактозидазы А и увеличение содержания Lyso-GL3 в высушенных каплях крови или плазме, а также наличие патогенной мутации гена GLA. При интерпретации результатов лабораторных тестов следует учитывать, что резкое снижение или полное отсутствие активности альфа-галактозидазы A в сочетании со значительным (в десятки раз) увеличением концентрации Lyso-GL3 наблюдается у мужчин с классическим вариантом заболевания, в то время как у женщин и пациентов с атипичным вариантом болезни Фабри активность лизосомного фермента может быть нормальной или близкой к норме, а концентрация Lyso-GL3 увеличивается в меньшей степени. Наличие мутации гена GLA служит обязательным критерием диагностики болезни Фабри, но самого по себе его недостаточно для установления этого диагноза, так как сегодня описано свыше 900 мутаций этого гена, а клиническое значение многих из них остается неизвестным. Полный набор критериев для установления достоверного диагноза болезни Фабри приведен в таблице 1 [2].

БОЛЕЗНЬ ГОШЕ (Е75.2)
Наиболее часто встречающаяся из лизосомных болезней – болезнь Гоше – характеризуется недостаточностью фермента глюкоцереброзидазы, которая вызывает накопление в организме больного жирного вещества, именуемого глюкоцереброзидом. Клетки-макрофаги с накоплением субстрата принято называть клетками Гоше. Накопление этих клеток в органах и костях способно привести к возникновению симптомов, которые могут варьировать от легкой до тяжелой степени и появляться в любом возрасте, от младенчества до зрелого периода [4].
Описаны 3 типа болезни Гоше, которые отличаются друг от друга главным образом степенью поражения центральной нервной системы:
- I тип – не нейронопатическая форма;
- II тип – острая нейронопатическая форма;
- III тип – хроническая нейронопатическая форма.
Особенности и различия этих трех форм болезни Гоше представлены в таблице 2.
Составляющими клинической картины болезни Гоше являются:
- гепатомегалия. Накопление клеток Гоше может также приводить к циррозу, образованию рубцов в печени и развитию печеночной дисфункции;
- спленомегалия:
- развивается гиперактивность органа;
- размеры селезенки могут увеличиться в 25 раз;
- развиваются анемия, тромбоцитопения, лейкопения;
- геморрагический синдром (носовые кровотечения, геморрагическая сыпь);
- костные боли (остеопороз, остеосклероз, асептический некроз);
- задержка физического и полового развития;
- гиперпигментация кожных покровов;
- поражение нервной системы.
При прогрессировании болезни может развиться портальная гипертензия, асцит, также поражаются другие органы – легкие, почки. Спектр поражений при болезни Гоше и их частота приведены на рисунке 3.

Диагностика болезни Гоше состоит из комплексной оценки клинической картины, лабораторных тестов и других исследований (например, ультразвукового исследования печени и селезенки), рентгенографии и компьютерной томографии (КТ) костей.
Из лабораторных исследований для диагностики болезни Гоше применяется ферментная диагностика: определение активности бета-глюкоцереброзидазы (уровень фермента снижен), хитотриозидазы (активность фермента повышена).
Генетическая диагностика включает секвенирование экзонов и приэкзонных участков интронов гена GBA методом ДНК-диагностики. При этом идентифицировано значительное количество мутаций гена GBA, приводящих к развитию болезни Гоше [5].
МУКОВИСЦИДОЗ (Е84)
Муковисцидоз – наследственное заболевание, при котором нарушаются функции желез внешней секреции. Ген муковисцидоза отвечает за контроль структуры и функции белка, который локализуется в апикальной части мембраны эпителиальных клеток, выстилающих выводные протоки желез внешней секреции (в бронхах, поджелудочной железе, кишечнике, урогенитальном тракте), и регулирует транспорт электролитов (главным образом хлора) между этими клетками и межклеточной жидкостью [6]. Мутации гена муковисцидоза нарушают не только транспорт, но и секрецию ионов хлора. При затруднении их прохождения через клеточную мембрану увеличивается реабсорбция натрия железистыми клетками, нарушается электрический потенциал просвета, что вызывает изменение электролитного состава и дегидратацию секрета желез внешней секреции. В результате выделяемый секрет становится чрезмерно густым и вязким. При этом страдают легкие, ЖКТ, печень, поджелудочная железа, мочеполовая система (табл. 3).

В некоторых случаях симптомы болезни проявляются не сразу. Это связано с тем, что тяжесть и форма заболевания у разных больных отличаются. Кроме того, признаки муковисцидоза могут напоминать симптомы других болезней, что затрудняет диагностику. Далее мы приведем признаки муковисцидоза у взрослых, так как детские формы заболевания чаще выявляются педиатрами.
У пациентов с «типичной» формой муковисцидоза, заболевших в раннем детстве и доживших до взрослого возраста, наблюдаются:
- низкий нутритивный статус;
- обструктивный тип вентиляционных нарушений;
- легочная гипертензия;
- постоянные рецидивы инфекционно-воспалительных процессов в легких при выраженных изменениях бронхиальной стенки;
- цирротические изменения;
- кровохарканье;
- пансинуситы;
- сахарный диабет.
Для атипичной формы муковисцидоза с поздней манифестацией характерны:
- синусит;
- рецидивирующий бронхит;
- хронические обструктивные болезни легких;
- цирроз печени;
- мужское бесплодие.
Особенности клинической картины муковисцидоза у взрослых:
- постоянный сухой кашель;
- трудноотделяемая гнойная вязкая мокрота;
- заложенность носа;
- гнойные выделения из носа;
- одышка;
- боли в животе;
- обильный, частый (4–6 раз в сутки), блестящий, жирный, зловонный стул;
- слабость;
- утомляемость;
- снижение массы тела/задержка в прибавке веса.
Характерный анамнез при муковисцидозе у взрослых:
- данные о смерти детей на первом году жизни;
- мекониальный илеус и его эквиваленты;
- синдром нарушенного кишечного всасывания неясного генеза;
- признаки хронического синусита с поражением всех синусов, полипы носа;
- рецидивирующие бронхиты, бронхиолиты;
- повторные и рецидивирующие пневмонии с затяжным течением;
- частые курсы антибактериальной терапии для лечения кашля;
- бронхиальная астма, рефрактерная к традиционной терапии;
- рецидивный панкреатит;
- цирроз печени;
- сахарный диабет с респираторным синдромом;
- гастроэзофагеальный рефлюкс;
- холелитиаз;
- выпадение прямой кишки;
- задержка полового развития.
При физикальном обследовании у взрослых пациентов с муковисцидозом наблюдается нарушение роста и развития, снижение массы тела. Отмечается типичный внешний вид: «кукольное» лицо, расширенная, деформированная грудная клетка бочкообразной формы с выбуханием грудины, большой, вздутый, иногда «лягушачий живот», худые конечности. Больные могут жаловаться на «соленый» вкус кожи. Также обнаруживаются признаки хронической гипоксии: деформации концевых фаланг пальцев по типу «барабанных пальцев» и/или ногтей по типу «часовых стекол». Перкуторно над легкими может выслушиваться коробочный звук и/или участки притупления, а при аускультации – ослабление дыхания, сухие и разнокалиберные (преимущественно среднепузырчатые) влажные хрипы – локальные или распространенные – в зависимости от объема поражения. При обследовании верхних дыхательных путей выявляется хронический пансинусит, нередко полипозный.
Схематично алгоритм диагностики муковисцидоза суммирован в таблице 4.

ГЕМОФИЛИЯ (D66.0, D67.0)
Наследственное заболевание свертывающей системы крови, возникающее в результате дефицита фактора свертывания крови VIII, обозначается как гемофилия А, а вследствие дефицита фактора свертывания крови IX – как гемофилия B. Распространенность гемофилии в большинстве стран составляет 10–14 больных на 100 000 мужчин. Гемофилия А встречается чаще, чем гемофилия В, и составляет 80–85% общего числа случаев этой болезни.
Гемофилия передается по X-сцепленному рецессивному пути наследования и поражает преимущественно мужчин. Представительницы женского пола являются носительницами патологического гена; случаи заболевания девочек во всем мире единичны [7].
Легкая форма гемофилии может никак не проявляться на протяжении всей жизни. Геморрагический синдром обычно возникает вследствие значительных травм или при хирургическом лечении. При гемофилии средней тяжести первые признаки развиваются после года в виде посттравматических гематом и длительных кровотечений (травма слизистых оболочек).
Наконец, тяжелая форма гемофилии сопровождается геморрагическим синдромом на первом году жизни (гематомы мягких тканей, посттравматические кровотечения из слизистых, гемартрозы).
Основные проявления гемофилии – кровотечения и кровоизлияния, возникающие спонтанно или вследствие травмы:
- кровоизлияния в кожу, мышцы и мягкие ткани;
- кровотечения из десен;
- кровотечения после операций;
- кровотечения после прививок;
- кровь в моче или стуле;
- кровотечения из носа;
- кровоизлияния в суставы.
Наиболее частые локализации кровотечений при гемофилии указаны на рисунке 4.

Повторяющиеся кровоизлияния в суставы (гемартроз) приводят к их необратимому повреждению (хроническая гемофилическая артропатия), а также к развитию:
- вторичных контрактур мягких тканей;
- мышечной атрофии;
- угловых деформаций конечностей (рис. 5).
Заподозрить гемофилию позволяет характерный семейный анамнез, а именно выявление факта геморрагического синдрома у членов семьи пациента. Геморрагические проявления у близких родственников по материнской линии (у мужчин, реже у женщин) отмечаются в семейном анамнезе примерно 2/3 больных гемофилией [8].
Наряду с этим, при диагностическом обследовании необходимо выяснить, наличие следующих «красных флагов» у самого пациента:
- легко появляющихся экхимозы и гематомы в раннем детстве;
- возникновение спонтанных кровотечений (особенно в суставы, мышцы и мягкие ткани);
- длительные кровотечения после травм или хирургического вмешательства;
- кефалогематома, внутричерепные кровоизлияния, кровоточивость и длительное заживлении пупочной ранки в неонатальном периоде;
- несоответствие выраженности геморрагических проявлений и тяжести предшествовавшей травмы;
- рецидивы кровотечений после их первичной остановки, не связанные с повторной травмой;
- массивные и (или) множественные гематомы;
- системность геморрагических проявлений (проявления различной локализации);
- «спонтанные геморрагические проявления».
При физикальном осмотре настороженность врача должны вызвать множественные экхимозы и гематомы, признаки поражения суставов (деформация, отек, локальное повышение температуры, нарушение подвижности).
Лабораторные диагностические исследования при подозрении на гемофилию включают:
1. Скрининговые анализы:
- определение активированного частичного тромбопластинового времени (АЧТВ; показатель почти всегда пролонгирован, однако у некоторых пациентов, с легкой формой гемофилии, его значение будет в пределах нормы);
- определение протромбинового времени;
- определение тромбинового времени;
- оценка концентрации фибриногена по Клауссу;
- измерение времени кровотечения стандартизованным методом;
- подсчет количества тромбоцитов по Фонио.
2. Оценку активности фактора VIII в крови – одностадийный или хромогенный анализы.
3. Генетический анализ: выполнение генотипирования для выявления конкретного генетического дефекта.
БОЛЕЗНЬ ПОМПЕ (Е74.0)
Болезнь Помпе – наследственное прогрессирующее заболевание, при котором в клетках из-за нехватки специфического фермента (кислой альфа-глюкозидазы) накапливается гликоген, постепенно нарушающий работу мышц и приводящий к мышечной дистрофии [9].
Выделяют три формы болезни:
- классическую с младенческим началом. Проявляется в течение нескольких месяцев после рождения, быстро прогрессирует и заканчивается летальным исходом от развития сердечно-легочной недостаточности или инфекций дыхательных путей, как правило, на первом году жизни;
- ювенильный вариант. Проявляется в детском или юношеском возрасте, протекает сравнительно мягко и поражает в основном скелетную мускулатуру;
- болезнь Помпе с поздним началом. Развивается в 20–50 лет, прогрессирует медленнее, чем остальные формы.
Дополнительно описана «кардиальная форма» заболевания, в тех случаях, когда ведущими клиническими симптомами выступают нарушения со стороны сердца.
Жалобы при болезни Помпе с поздним началом:
- дневная сонливость;
- изменение походки (пациент ходит «вперевалку»);
- неустойчивость при ходьбе (пациент может упасть, если нагнется вперед);
- эпизоды головокружения при перемене положения тела;
- трудности при вставании с низкого стула;
- одышка при разговоре и ходьбе;
- изменение артикуляции (неразборчивая речь);
- подъем из положения лежа на спине возможен только при опоре на руки;
- трудности при натуживании (процесс дефекации занимает не менее 40 мин);
- учащенное мочеиспускание;
- снижение слуха;
- периодические боли в ногах;
- утренняя головная боль.
Ретроспективный анализ жалоб пациентов, страдающих болезнью Помпе с поздним началом, показал, что первые проявления или признаки заболевания, которые субъективно не рассматривались пациентом как отклонение от нормы, в 77% случаев касались трудностей при занятиях спортом (пациенты плохо бегали, не могли подтянуться на турнике), в 28% – быстрой усталости при ходьбе по лестнице, в 20% – затруднений при вставании с низкого стула, в 17% – усталости при обычной ходьбе и в 11% – неспособности встать из положения лежа на спине без помощи рук [9].
При осмотре пациента крайне важно обратить внимание на следующие симптомы.
1. Слабость мышц передней стенки живота.
2. Слабость сгибателей шеи.
3. Слабость параспинальных мышц (лордоз, кифоз, сколиоз).
4. Слабость ягодичных мышц и трудность удержания равновесия при вставании.
5. Сохранность силы четырехглавых мышц при слабости мышц других отделов.
6. Слабость диафрагмы, одышка.
7. Слабость мышц языка.
8. Слабость мимических мышц.
9. Утренняя головная боль, тахикардия, дневная сонливость, необычная утомляемость.
Диагностика болезни Помпе с поздним началом осложнена тем, что жалобы пациента абсолютно неспецифичны и связаны в основном со слабостью скелетной мускулатуры, что можно списать на естественное старение организма. Изменения со стороны мышечной, сердечно-сосудистой и дыхательной систем могут быть минимальны, а больной длительное время остается активным, с неизменным образом жизни. Однако, неуклонное прогрессирование симптомов и значительное снижение качества жизни пациента могут помочь заподозрить данное орфанное заболевание.
Диагностика болезни Помпе включает биохимический анализ крови с определением уровня сывороточной креатинкиназы (КФК), аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), лактатдегирогеназы (ЛДГ). Рекомендуется оценка активности КАГ (кислой мальтазы) в пятнах крови, высушенной на фильтровальной бумаге, лейкоцитах периферической крови. Этот тест сочетает высокую чувствительность с простотой выполнения и возможностью применять его для скрининга большого числа пациентов. Определение активности КАГ в сухом пятне крови является оптимальным скрининговым тестом, «золотым стандартом», позволяющим малоинвазивно, быстро и точно определить дефицит активности фермента [10].

ЗАКЛЮЧЕНИЕ
Таким образом, многие из проявлений орфанных заболеваний не относятся к патогномоничным и/или специфическим. Все это требует известной настороженности и осведомленности врачей терапевтов и врачей общей практики в отношении таких патологий. В таблице 5 обобщены самые распространенные и яркие проявления орфанных заболеваний, помогающие в их диагностике.
