
Новейшие достижения в исследованиях позволяют ставить гипертрофическую кардиомиопатию (ГКМП) в ряд наиболее распространенных (0,2% в общей популяции) наследственных сердечно‑сосудистых заболеваний (ССЗ) [1]. Наследуясь по аутосомно‑доминантному типу, ГКМП может поражать несколько индивидов в семейной родословной из-за нарушения продукции структурных, ферментных или регуляторных белков саркомера кардиомиоцита. Поэтому ГКМП все чаще характеризуют как «саркомеропатию» или «саркомерную кардиомиопатию» [2]. Уже сформировано представление о специфической морфологической матрице заболевания [3]. Она обычно реализуется в основные механизмы нарушения диастолической функции, аритмии, сердечной недостаточности (СН) или ишемии гипертрофированного миокарда. Степень превалирования одного или нескольких из этих механизмов определяет клиническую гетерогенность и исход заболевания во внезапную смерть (ВС), в СН (вплоть до формирования рефрактерной СН — «конечная стадия») или долговременное благоприятное течение («безмолвные» формы ГКМП) [1, 2, 5, 7–9, 12–15].
Достижения в молекулярно‑генетических исследованиях подтвердили генетическую гетерогенность ГКМП и роль различных генных мутаций в формировании разновидных фенотипов заболевания [1, 5, 9–11, 14, 18, 22]. Сходные генетические дефекты саркомерных белков вследствие de novo аллельных мутаций выявлены в 20–40% случаев заболеваний (так называемые спорадические формы ГКМП), по данным разных исследователей [1, 9, 10, 13, 15,17–19]. В 50% случаев эти дефекты пенетрируют в следующих поколениях. Новейшие результаты подкрепляют ранее возникшие предположения о значимых корреляциях между выявленными генетическими дефектами и исходом заболевания [1,13, 14, 17–20, 22]. Однако наряду с этим ряд исследователей сообщает о сходных морфофункциональных фенотипах и исходах заболеваний, имевшихся при разных мутациях в одной и той же линии наследственности, а также о различных фенотипах при сходных мутациях в одном и том же гене саркомерных белков [1, 3, 10, 17–19]. Имеются подтвержденные предположения о значении внешних факторов (таких как метаболические или воспалительные) или других мутаций «модифицирующих генов», не принадлежащих к специфичному генотипу ГКМП, но воздействующих на формирование фенотипа [1, 2, 5, 16–19, 23]. Пока не удается выявить четкие корреляции между разновидным генотипом и фенотипом ГКМП, что и является значительным барьером на пути разработок и внедрения эффективного наследственного скрининга для стратификации рисков и прогнозирования течения заболеваний.
Для дальнейшего продвижения исследований требуется внедрение эффективных и всеобъемлющих инструментов клинического и наследственного скрининга [1, 5, 6, 14, 19–22] для полномерной оценки возможных профилей фенотипирования. Изучение клинических профилей наследственной ГКМП и являлось целью представленного исследования.
Материал и методы
При обращении в клинику обследованы 64 мужчины и женщины с ГКМП, средний возраст которых к моменту включения в исследование составлял 43,8±11,7 года. Предварительную оценку случаев ГКМП осуществляли по данным стандартного протокола трансторакальной эхокардиографии (ЭхоКГ). Проводили дифференциальную диагностику с другими сердечно‑сосудистыми или системными заболеваниями, которые могли стать причиной асимметричной или симметричной гипертрофии миокарда (ГМ). Во всех выявленных случаях ГКМП выполняли скрининговое обследование родственников пробанда I степени родства и родственников старшего и младшего поколения. Выявленных пациентов с ГКМП включали в исследуемую группу.
На основании изучения семейного анамнеза при помощи членов семьи и анализа существующих медицинских записей проводили ретроспективную оценку обстоятельств смерти и определяли возможность случаев ВС по основным критериям: 1) состояние пациента до смерти определяли как стабильно удовлетворительное; 2) смерть наступила в течение часа после появления острых симптомов заболевания или не позже 6 ч после того, как пациента видели живым; 3) смерть не могла быть вызвана другой причиной, кроме заболевания [24].
Тяжесть хронической СН оценивали по классификации функциональных классов (ФК) Нью‑Йоркской ассоциации сердца, а также по стадиям с использованием показателей диастолической и систолической функции сердца [29]: 1) компенсированная — нарушение пассивной диастолической функции; 2) нарушение пассивной и активной диастолической функции; 3) декомпенсированная — развернутая диастолическая и систолическая дисфункции.
По данным электрокардиографии в 12 стандартных отведениях изучали критерии Соколова—Лайона, глубину инверсии зубца Т и депрессию сегмента ST в 2 отведениях и более, за исключением неинтерпретируемых случаев (постоянная форма мерцания предсердий, полная блокада левой ножки пучка Гиса). Величины корригированного интервала Q—T (Q—Tc) и его дисперсии (Q—Tcd) определяли по методу Glancy [25]. При суточном мониторировании электрокардиограммы (ЭКГ) выявляли число желудочковых экстрасистол (ЖЭ) и эпизодов нестойкой желудочковой тахикардии (ЖТ).
При ЭхоКГ определяли основные показатели, характеризующие структурные и функциональные изменения сердца: индекс толщины межжелудочковой перегородки (ТМЖП); индекс толщины задней стенки левого желудочка (ТЗСЛЖ); относительную толщину стенок левого желудочка (ЛЖ); индекс конечного диастолического объема ЛЖ; индекс массы миокарда ЛЖ; индекс площади поверхности левого предсердия; фракцию выброса ЛЖ [26, 27, 29]. Оценку диастолической функции ЛЖ осуществляли в допплеровском режиме по данным времени изоволюмической релаксации (ВИР) ЛЖ и индекса (Е/А) трансмитрального кровотока в фазе наполнения ЛЖ.
Статистическую обработку полученных данных проводили с использованием статистической программы для IBM Minitab 11 for Windows. Для анализа количественных данных использовали t‑критерий, непараметрический критерий Манна—Уитни для малых и неравных выборок, коэффициент корреляции по отдельным выбранным показателям. Различия считались статистически значимыми при p<0,05.
Результаты
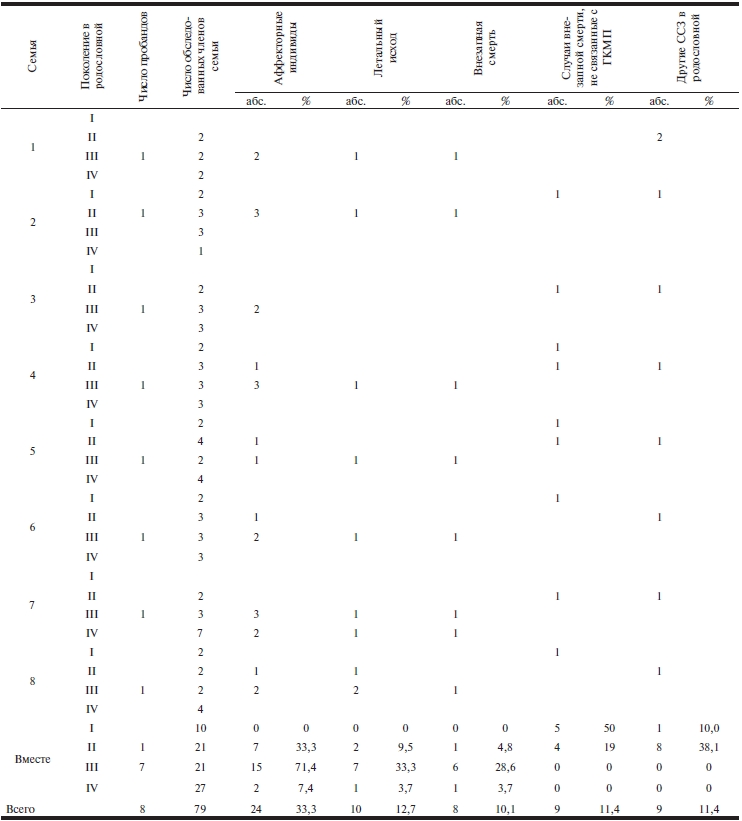
При исследовании родословной 40 (62,5%) пациентов новые случаи заболевания у родственников I степени родства не выявлены. Эти случаи были расценены как спорадические формы ГКМП. У 8 пациентов‑пробандов в родословной выявлены дополнительно 16 пациентов. Таким образом, наследственный скрининг позволил выявить 24 (37,5%) случая семейных форм ГКМП (табл. 1, 2). Для полномерного скрининга в 8 семьях пробандов были обследованы 79 лиц, принадлежащих четырем поколениям родословной. Наибольшее число аффекторных пациентов были выявлены в III поколении — 15 (71,4%) из 21 обследованного члена семьи пробандов. Из 8 выявленных на предварительном этапе скрининга 7 пробандов принадлежали именно этому поколению. Умерли 1/3 обследованных в этом поколении и в большинстве случаев в связи с ВС — 6 (28,6%) из 21. При изучении II поколения родословной выявлены 1/3 аффекторов — 7 (33,3%) из 21. Летальный исход был зафиксирован в 2 (9,5%) случаях, в одном из них — ВС (4,2%). При наследственном скрининге в IV поколении родословных обнаружены 2 (7,4%) аффектора из 27 обследованных, из них умер один (3,7%). Выявлены 5 случаев ВС, не связанных с ГКМП, из 10 обследованных членов семей I поколения (50%). В этом поколении не было ни одного выявленного случая ГКМП. Во II поколении среди 21 обследованного определены 4 (19%) случая ВС, не связанные с ГКМП. По результатам исследования родословных всех 8 пробандов клинический фенотип ГКМП был выявлен у 30,2% среди 79 родственно связанных индивидов, умерли 12,7%. У 10,1% от общего числа обследованных по родословному ряду была ВС.
Таблица 1. Фенотипирование заболеваний в семейной родословной
Примечание. ГКМП — гипертрофическая кардиомиопатия; ССЗ — сердечно-сосудистые заболевания.
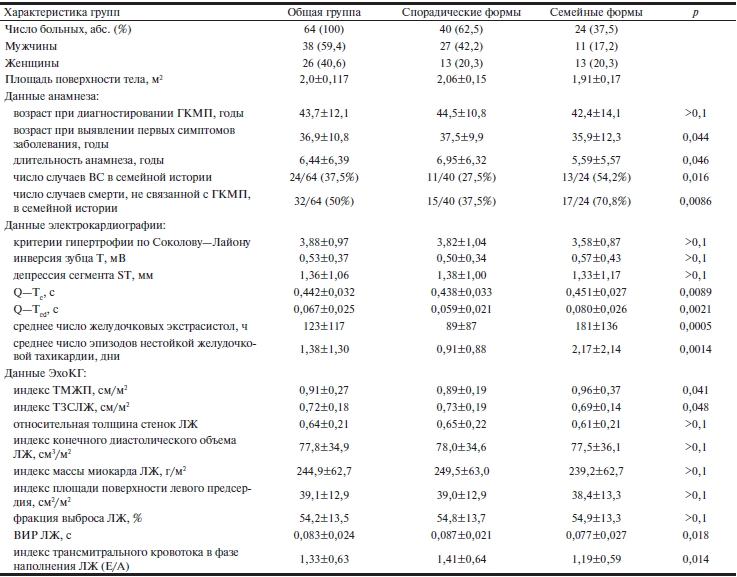
Таблица 2. Основные данные обследования больных по группам
Примечание. ГКМП — гипертрофическая кардиомиопатия; ВС — внезапная смерть; ТМЖП — толщина межжелудочковой перегородки; ТЗСЛЖ — толщина задней стенки левого желудочка; ЛЖ — левый желудочек; ВИР — время изоволюмической релаксации.
В исследуемых группах пациентов со спорадическими и семейными формами ГКМП благоприятное течение было констатировано в 65 и 58,3% случаев соответственно. Высокая летальность определялась в группе пациентов с семейными формами ГКМП (рис. 1). Частота ВС была значительно выше в этой группе (33,3% против 15%). В группе пациентов со спорадическими формами преобладали летальные исходы, связанные с «конечной стадией» заболевания (22,5% против 8,3%).

Рис. 1. Исход при спорадических и семейных форм гипертрофической кардиомиопатии.
При изучении анамнеза пациентов также была выявлена высокая частота случаев ВС (13 из 24, или 54,2%) и летальных исходов в связи с другими ССЗ (17 из 24, или 70,8%) в семейной истории пациентов с наследованными формами ГКМП по сравнению с пациентами в группе со спорадической ГКМП.
В общей группе обследованных больных преобладали случаи хронической СН III—IV ФК (рис. 2), такая же тенденция определялась и в группе пациентов со спорадической ГКМП. В группе семейной ГКМП преобладали пациенты с хронической СН I—II ФК (58,3% против 41,7% с III—IV ФК).

Рис. 2. Оценка сердечной недостаточности в исследуемых группах по классификации Нью Йоркской ассоциации сердца.
При оценке степени ГМ по данным ЭКГ достоверные различия не выявлены. Электрокардиографические критерии по Соколову—Лайону, глубина инверсии зубца Т, депрессии сегмента ST не различались у пациентов со спорадическими или семейными формами ГКМП (см. табл. 2). Интервал (Q—Tc) у пациентов с семейными ГКМП оказался достоверно шире (0,451±0,027 с), чем у пациентов со спорадическими формами (0,438±0,033 с). Выявлено значимое различие и дисперсии интервалов Q—T (Q—Tcd) для пациентов с семейными и спорадическими формами ГКМП (0,080±0,026 с против 0,059±0,021 с). Эти показатели коррелировали с частотой выявления ЖЭ и эпизодов ЖТ (коэффициенты корреляции 0,880 и 0,885 соответственно), особенно в группе пациентов с семейными формами заболевания. По данным суточного мониторирования ЭКГ в группе с семейной ГКМП были выявлены достоверно частые ЖЭ (181±136 ч–1) и эпизоды ЖТ (2,17±2,14 в сут–1) по сравнению с таковыми в группе пациентов со спорадическими формами (89±87 ч–1 и 0,91±0,88 в сут–1 соответственно).
По данным ЭхоКГ также выявлены некоторые существенные различия (см. табл. 2). В группе с семейными формами ГКМП достоверно выше оказались показатели ТМЖП (0,96±0,37 см/м2 по сравнению с 0,89±0,19 см/м2 в группе со спорадическими формами), достоверно ниже — ТЗСЛЖ (0,69±0,14 см/м2 по сравнению с 0,73±0,19 см/м2). Расчеты показателей конечного диастолического объема, относительной толщины миокарда, индексов объема полости и массы миокарда ЛЖ, индекса площади поверхности левого предсердия не выявили достоверных различий между исследуемыми группами. Фракция выброса ЛЖ также не различалась в группах пациентов со спорадическими и семейными формами ГКМП (54,8±13,7 и 54,9±13,3% соответственно).
При оценке диастолической функции обнаружены достоверное увеличение ВИР при спорадических формах ГКМП (0,087±0,021 с по сравнению с 0,077±0,027 с при семейных формах) и достоверное увеличение отношения показателей раннего быстрого и предсердного медленного наполнения (Е/А) в группе со спорадическими формами (1,41±0,64 по сравнению с 1,19±0,59 в группе с семейными формами ГКМП). Выявлены различные виды нарушения функции ЛЖ: в группе пациентов с семейными формами ГКМП достоверно преобладали случаи I и II стадии хронической СН, тогда как в группе со спорадическими формами, как и в общей группе, в равней степени выявлялись все три стадии (рис. 3).

Рис. 3. Стадии сердечной недостаточности с учетом формы ГКМП.
Обсуждение
Результаты данного исследования подтвердили роль наследования в фенотипировании ГКМП. В нашем исследовании при клиническом скрининге в 37,5% случаев выявлена наследственная обусловленность заболевания. Примерно такая же частота (30,2%) выявлена при клиническом скрининге в родословных пробандов. В ряде последних научных сообщений указывается на наследование различных заболеваний в 60–80% случаев [1, 3, 14, 15, 19–22]. Но в этих исследованиях результаты основываются на результатах молекулярно‑генетического скрининга и по сути речь идет о наследственной передаче измененного генотипа, а не только клинического фенотипа. Вместе с этим выявляются случаи измененных генотипов, которые не реализуются в клинический фенотип в родословной, и случаи, когда мутации «стираются» в последующих поколениях.
В 5 (62,5%) семьях из 8 наши исследования по необходимым мероприятиям наследственного скрининга затронули 4 поколения родословной и аффекторные пациенты были идентифицированы в 3 поколениях. Более уязвимым оказалось поколение пробандов, в котором было выявлено наибольшее количество клинических фенотипов родословной (75% от общего числа случаев семейных форм), и именно в этом поколении были зафиксированы летальные исходы, связанные с ГКМП (37,5%) с превалированием ВС (33,3%). При анализе данных по группам исследования ВС оказалась достоверно частым исходом при семейных формах ГКМП, а так называемая конечная стадия заболевания — при спорадических формах. Результаты новейших исследований связей генотипов и фенотипов ГКМП отражают роль наследственности в передаче отдельных признаков заболевания, в том числе случаев ВС как наследуемого признака. Результаты нашего исследования согласуются с этими предположениями [1, 3, 10, 11, 15, 17–22]. Наши суждения о возможном наследовании генетических дефектов, формирующих морфологический и физиологический субстрат для развития исходов в виде ВС, подтверждаются и другими результатами исследования. Случаи ВС, прямо не связанные с фенотипом ГКМП, достоверно чаще выявлялись в группе семейных форм (54,2%), чем в группе спорадических форм заболевании или в общей группе. Возможные профили рисков в виде случаев ВС в предыдущих поколениях наблюдались нами как «агрессивный» признак и в ранних клинических исследованиях [5–8].
Обсуждая результаты ЭКГ, можно отметить, что признаки ГМ существенно не различались в исследуемых группах. В группе с семейной ГКМП выявлялись нарушения электрической актив‑ ности сердца: показатели дисперсии процессов возбуждения и рефрактерности миокарда в виде удлиненного интервала Q—Т и значительной его изменяемости. Эти нарушения, обычно обусловленные специфическими морфологическими изменениями при ГКМП (коэффициент корреляции 0,893 и 0,821), реализуются в «потенциально злокачественные» желудочковые аритмии, что и наблюдалось нами достоверно чаще среди пациентов с семейными формами ГКМП. Данные суточного мониторирования ЭКГ отражали частые ЖЭ и эпизоды ЖТ в этой группе пациентов. В настоящее время причинно‑следственные связи этих нарушений в развитии ВС приняты для клинической модели ГКМП и других ССЗ [1, 3, 7, 17–20] и выделены как ранние прогностические факторы риска.
По данным ЭхоКГ выявлено достоверное увеличение индекса ТМЖП и уменьшение индекса ТЗСЛЖ при семейной ГКМП. Полученные результаты указывают на более высокую степень и асимметричное распространение ГМ при этих формах заболевания.
Описанные выше структурные изменения формируют первичные нарушения пассивного компонента диастолической функции ЛЖ. Исследование также выявило более глубокие нарушения компонентов диастолы. Семейные формы заболевания характеризовались достоверно низким показателем индекса трансмитрального кровотока (Е/А) и ВИР ЛЖ, что указывает на нарушения в начальной стадии диастолы, в фазе быстрого наполнения ЛЖ и превалирующем пассивном компоненте диастолы. При спорадических формах данные допплерографии отражают более глубокое нарушение активной диастолы, удлинение ВИР и фазы медленного наполнения ЛЖ. Эти функциональные нарушения при ГКМП описываются в разных клинических исследованиях [1, 3, 8, 9, 11–14, 26–28]. Полученные данные, конечно, невозможно прямо связать с какими‑либо специфичными генетическими нарушениями и представить как наследуемый признак. Однако о таких результатах исследовании сообщается разными авторами. При этом в обусловленности диастолической дисфункции нельзя не учитывать роль возможных внешних факторов. Имея прямую причинно‑следственную связь со специфическими структурными нарушениями, передаваемыми по наследству, они представляют собой важную составляющую фенотипа ГКМП.



{kind=link}
{kind=link}