
В настоящее время смертность и инвалидность от осложнений инфаркта миокарда (ИМ) остаются высокими, что определяет необходимость повышения эффективности прогнозирования неблагоприятного течения заболевания. Доказано, что пациентам с высоким риском развития осложнений требуется дорогостоящее лечение, тогда как в группе с низким риском лечение может быть более избирательным, что позволяет более эффективно распределять ресурсы общества [1].
Одной из наиболее простых и получивших широкое распространение систем оценки риска развития осложнений ИМ является индекс TIMI Risk Score-STEMI [2]. Данный метод позволяет оценить вероятность летального исхода ИМ в течение 30 дней от начала заболевания у пациентов, подвергшихся тромболитической терапии (ТЛТ). Это единственная шкала стратификации риска, одобренная Российским кардиологическим обществом для практического применения у больных ИМ с подъемом сегмента ST (ИМпST). Несмотря на простоту использования, она обладает достаточной высокой прогностической ценностью — площадь под ROC-кривой при использовании данной системы на целевой выборке пациентов составляет 0,78 [2].
Относительно недавно в кардиологическую практику вошла шкала GRACE (Global Registry of Acute Coronary Events) [3], построенная с использование выборки, включающей 43 810 пациентов. Данная модель продемонстрировала хорошие прогностические возможности: показатель С-статистики в отношении стратификации риска смерти в течение 6 мес составил 0,81, а госпитальной летальности — 0,82, что подтвердилось при проверке данной модели на базе данных регистра GUSTO IIb [3]. Данная модель — единственная, которая позволяет оценить риск не только смерти, но и рецидива нефатального ИМ, и применима для всех больных вне зависимости от метода реперфузии миокарда. С целью определения степени индивидуального риска по шкале GRACE разработана программа, on-line версия которой представлена на сайте http://www.outcomes.org.

Несмотря на существование нескольких альтернативных способов оценки риска развития осложнений ИМ, для практикующего врача остается открытым вопрос о выборе наиболее чувствительного и специфичного из них. Кроме того, необходимо наличие возможности выбора прогностического подхода в зависимости от демографических, социальных и экономических условий конкретной популяции. Недостатком существующих методов является также и то, что они не учитывают генетические факторы, которые играют важную роль в структуре подверженности сердечно-сосудистым осложнениям (ССО) [4] и остаются постоянными на протяжении жизни, т.е. их оценка возможна на любом этапе, в том числе задолго до манифестации заболевания (предиктивное тестирование).
В исследовании S.E. Humphries и соавт. [5] получены доказательства того, что система оценки риска развития ССО в общей популяции, включающая наряду с традиционными факторами риска (ФР) набор нескольких генотипов, была более эффективна, чем система, основанная исключительно на традиционных ФР. Работа была выполнена с использованием данных Northwick Park Heart study II (Великобритания), где наблюдение проводилось за выборкой, включающей 3052 мужчин среднего возраста, за 10-летний период было зафиксировано 250 коронарных осложнений. Точность диагностики традиционной системы оценки риска и оценки риска, основанной исключительно на генетических маркерах, оказались сопоставимыми, а комбинация генетических и традиционных ФР улучшала показатель системы примерно на 10%.
Кроме того, получены данные о том, что генетические маркеры могут повышать прогностическую способность шкал, предназначенных для прогнозирования риска развития осложнений ИМ. Включение в модель GRACE информации о генотипе по полиморфному варианту rs1333049, расположенному в локусе хромосомы 9р21, позволяло улучшить ее прогностическую значимость на 5,9% и тем самым точнее предсказывать принадлежность пациентов к той или иной группе риска развития повторного ИМ/ смерти [6].
Целями настоящего исследования являлись построение модели оценки риска развития осложнений ИМ, основанной на анализе полиморфных генетических вариантов генов-кандидатов сердечно-сосудистых заболеваний (ССЗ) и анализ ассоциаций индивидуальных генетических маркеров со статусом пациентов через 12 мес после острого заболевания.
Материал и методы
 В исследование включены 165 пациентов, госпитализированных в Кемеровский кардиологический диспансер по поводу острого коронарного синдрома с подъемом сегмента ST давностью менее 24 ч. Алгоритм обследования включал сбор жалоб, анамнеза, клинический осмотр кардиологом, запись электрокардиограммы, оценку уровня кардиоспецифических ферментов, контроль показателей системной гемодинамики, проведение эхокардиографии. Всем пациентам в кратчайшие сроки определяли предпочтительный метод реперфузии миокарда — чрескожное коронарное вмешательство (ЧКВ) или системную ТЛТ. Реваскуляризацию миокарда не проводили при наличии технических ограничений вследствие анатомии коронарных сосудов, противопоказаний к ТЛТ или ЧКВ.
В исследование включены 165 пациентов, госпитализированных в Кемеровский кардиологический диспансер по поводу острого коронарного синдрома с подъемом сегмента ST давностью менее 24 ч. Алгоритм обследования включал сбор жалоб, анамнеза, клинический осмотр кардиологом, запись электрокардиограммы, оценку уровня кардиоспецифических ферментов, контроль показателей системной гемодинамики, проведение эхокардиографии. Всем пациентам в кратчайшие сроки определяли предпочтительный метод реперфузии миокарда — чрескожное коронарное вмешательство (ЧКВ) или системную ТЛТ. Реваскуляризацию миокарда не проводили при наличии технических ограничений вследствие анатомии коронарных сосудов, противопоказаний к ТЛТ или ЧКВ.
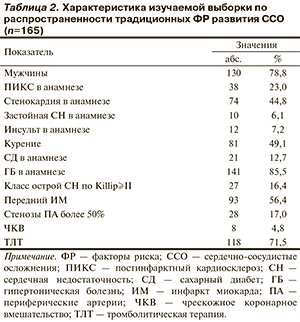
В табл. 1 представлены средние значения некоторых клинически значимых показателей и оценка риска по шкалам GRACE и TIMI в обследованной выборке больных. Так, средний возраст больных составил 59 лет, средняя оценка по шкале GRACE — 98,3 балла, по шкале TIMI — 2,9 балла. В табл. 2 приведены данные о распространенности традиционных ФР развития ССО в исследованной выборке больных.
У всех пациентов определены генотипы по 32 полиморфным вариантам 24 важнейших генов-кандидатов ССЗ (табл. 3). Генотипирование проводили с помощью ДНК-чипа СИНКАР-1 (разработчики НИИ медицинской генетики СО РАМН и ООО «Геномная диагностика»). Использованный метод основан на технологии минисеквенирования (однонуклеотидного удлинения иммобилизированных праймеров). Процесс определения генотипов включал предварительную ПЦР-амплификацию исследуемых локусов (32 ПЦР-продукта); обработку щелочной фосфатазой и урацил-гликозилазой с целью фрагментации и удаления неиспользованных dNTP; проведение гибридизации и реакции однонуклеотидного удлинения праймеров на чипе (инкубация с флуоресцентно мечеными дидезоксинуклеозидтрифосфатами и ферментом секвеназой); отмывку реагентов с чипа; сканирование чипа в специальном сканере; анализ изображения и чтение результатов [7]. Чипы и реактивы для минисеквенирования, а также сканер чипов произведены в Эстонии. Приготовление смеси для реакции минисеквенирования осуществляли согласно протоколу производителя.
Данные о статусе пациентов через 1 год (жив или умер) были определены у 165 больных. Зафиксировано 8 смертельных исходов, все от ССО (4,8% от общего числа обследованных пациентов). Подробная клиническая картина, в том числе наличие других «конечных точек» и объем сопутствующей терапии получены для 146 (88,5% от всех включенных в исследование) больных. Данная подгруппа была в дальнейшем использована для построения генетической шкалы оценки риска развития осложнений ИМ. За «конечные точки» наблюдения принимали смерть через год после острого ССО (зафиксирована у 8 пациентов) и наличие нефатального ИМ (зафиксирован у 10 пациентов). Общая частота развития исходов, принятых за «конечную точку», составила 12,3%.
Статистическую обработку данных осуществляли с помощью программ SPSS версии 16.0 и JMP версии 8. Для анализа различий частот генотипов и аллелей использовали критерий χ2 Пирсона или точный тест Фишера при малом количестве наблюдений в какой-либо из подгрупп. Различия считали статистически значимыми при р<0,05.

Для построения модели прогнозирования осложнений после острого ИМ (повторного острого нефатального ИМ или смерти пациента в течение года) выбран прямой пошаговый алгоритм регрессионного анализа выживаемости по Коксу в отношении всех включенных в исследование генетических вариантов. Применение пошагового метода позволяет выбирать из некоторого количества факторов (генотипов) те, которые вносят наиболее существенный вклад в вариацию изучаемой переменной («конечной точки» — статуса пациента через год после острого коронарного осложнения). При пошаговом подходе факторы (индивидуальные генотипы) последовательно включаются в уравнение регрессии и проверяется их значимость. Если при включении в модель какого-то факторного признака величина множественного коэффициента корреляции увеличивается, а коэффициент регрессии меняется несущественно, то данный признак (генетический вариант) существенен и его включение в уравнение регрессии является необходимым.
Результаты и обсуждение
При построении модели прогнозирования риска анализировали 32 полиморфных варианта, имеющих отношение к различным механизмам формирования ССЗ (см. табл. 3). Данный набор генетических маркеров (генетический тест) реализован в формате биочипа и содержит в основном многократно подтвержденные (реплицированные в независимых исследованиях) генетические маркеры. В основу теоретической разработки данного генетического теста заложены современные представления о патогенезе ССЗ, включая понятие о сердечно-сосудистом континууме (непрерывности в цепи развития ССЗ от воздействия ФР до общего финального исхода в виде сердечной недостаточности и смерти), коморбидности, синтропии и плейотропных эффектах некоторых генов [8—11].
Лучшая по прогностической силе модель включала 3 генетических маркера. Из них 2 имели отношение к регуляции свертывания крови (rs6025 F5 и rs5918 ITGB3), третий — вариант в гене ангиотензинпревращающего фермента — АПФ (rs4291 ACE), имеющего чрезвычайно широкий спектр действия на сердечно-сосудистую систему. Носительство «рисковых» генотипов по этим генам позволяет с достаточной точностью прогнозировать развитие неблагоприятного исхода (смерти или нефатального ИМ) в течение 12 мес (табл. 4).
На рис. 1 представлена ROC-кривая полученной модели. Значение C-статистики составило 0,75 (0,64; 0,86) при р=0,001. При этом, как показано нами ранее [12], для шкалы GRACE для изучаемой выборки больных этот показатель составил 0,73 (0,61; 0,85). Таким образом, модель, включающая только генетические ФР, была сопоставима по прогностической силе с моделью, используемой в настоящее время в клинической практике, и даже несколько превосходила ее. Дальнейшее подтверждение клинической ценности полученной модели прогнозирования осложнений после ИМ требует анализа независимых выборок и проспективной оценки.
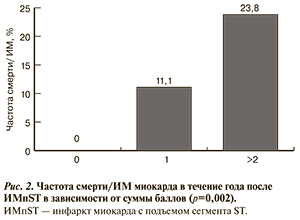
 Для удобства практического использования разработан алгоритм балльной оценки риска последующих неблагоприятных методов в зависимости от количества «рисковых» генотипов. Так, генотипу ТТ полиморфизма rs4291 гена АСЕ присваивается 2 балла, наличие гетерозиготного генотипа АТ оценивается в 1 балл, а генотип АА — 0 баллов. Генотипам GA и AA полиморфизма rs6025 гена F5 присваивается 1 балл и генотипу GG — 0 баллов. Генотипы СТ и СС полиморфизма rs5918 гена IGTB3 также оцениваются 1 баллом, а генотип ТТ этого гена — 0 баллов. При расчете смертности в течение года в подгруппе пациентов, у которых общая оценка составляет 0 (n=33), не зафиксировано ни одного смертельного исхода; в подгруппе с оценкой 1 балл — 2,6% (2 пациента из 76); с оценкой 2 балла и более — 12,2% (6 из 49). Частота фиксации выбранной в данном исследовании «конечной точки» (смерть/ИМ) при 0 баллов составила 0 (n=30), при 1 балле — 11% (8 из 73), при 2 баллах и более— 23,8% (10 из 42) (рис. 2).
Для удобства практического использования разработан алгоритм балльной оценки риска последующих неблагоприятных методов в зависимости от количества «рисковых» генотипов. Так, генотипу ТТ полиморфизма rs4291 гена АСЕ присваивается 2 балла, наличие гетерозиготного генотипа АТ оценивается в 1 балл, а генотип АА — 0 баллов. Генотипам GA и AA полиморфизма rs6025 гена F5 присваивается 1 балл и генотипу GG — 0 баллов. Генотипы СТ и СС полиморфизма rs5918 гена IGTB3 также оцениваются 1 баллом, а генотип ТТ этого гена — 0 баллов. При расчете смертности в течение года в подгруппе пациентов, у которых общая оценка составляет 0 (n=33), не зафиксировано ни одного смертельного исхода; в подгруппе с оценкой 1 балл — 2,6% (2 пациента из 76); с оценкой 2 балла и более — 12,2% (6 из 49). Частота фиксации выбранной в данном исследовании «конечной точки» (смерть/ИМ) при 0 баллов составила 0 (n=30), при 1 балле — 11% (8 из 73), при 2 баллах и более— 23,8% (10 из 42) (рис. 2).
В некоторых теоретических работах получены доказательства возможности использования ограниченного набора распространенных полиморфных генетических вариантов с небольшим индивидуальным вкладом для объяснения значительной доли «груза» заболевания в популяции. Так, S.E. Humphries и соавт. [5] рассчитали, что для улучшения прогностической способности существующей в кардиологии системы оценки риска развития ССО необходимы всего 12 SNP (single nucleotide polymorphism — однонуклеотидный полиморфизм) с частотой «рискового» генотипа 10% (при отношении шансов — OШ=1,5) или 3 SNP с частотой в популяции 30% для аналогичного увеличения прогностической силы. Сделан вывод о том, что небольшое количество полиморфных генетических вариантов будет достаточным для идентификации индивидов с высоким риском развития заболевания. В настоящем исследовании эффективная модель оценки риска развития осложнений ИМ построена с вовлечением только 3 генетических маркеров.
В дальнейшем проводили сравнение частот аллей и генотипов в подгруппах больных с благоприятным и неблагоприятным исходами и расчет ОШ. В табл. 5 приведены частоты аллелей и генотипов и ОШ для редкого аллеля в тех случаях, когда были зафиксированы статистически значимые различия по частотам генотипов между подгруппами больных с благоприятными и неблагоприятными исходами, а также характеристика вариантов, вошедших в модель оценки риска развития осложнений ИМ как независимых предикторов.
Ген F5 кодирует белок пятого фактора свертывания крови. Полиморфизм rs6025, известный как фактор Лейдена (лейденская мутация), заключается в замене гуанина (G) аденином (A) в положении 1691 (1691 G>A) последовательности нуклеотидных оснований, что приводит к замене аминокислоты аргинин глутамином в 506-й позиции белка (R506Q). Такая замена клинически проявляется повышенной свертываемостью крови и склонностью к тромбозам [13]. Имеются данные о том, что этот генетический вариант ассоциирован с высоким риском развития венозных тромбоэмболий и ишемического инсульта [14, 15]. В двух мета-анализах показано увеличение риска развития ИМ в 1,3 раза в случае выявления лейденской мутации [16, 17], однако данные противоречивы [13, 18]. При анализе подгрупп больных с благоприятным и неблагоприятным годовыми исходами (см. табл. 5), показано что в выборке с осложнениями в виде повторного нефатального ИМ или смерти чаще встречаются носители патологического аллеля А (лейденской мутации), ОШ для аллеля А составило 15,875 (p=0,0037) (см. табл. 5). ОШ для гомозигот по лейденской мутации при сравнении с гомозиготами по нормальному аллелю (GG) составило 24,484 (p=0,0049), OШ для гетерозиготного генотипа AG составила 8,400 при 95% ДИ от 1,101 до 64,071 (p=0,0158). Таким образом, в настоящем исследовании показана роль лейденской мутации как ФР развития осложнений ИМ в виде рецидива ИМ/смерти в течение года.

Вариант rs4291 (А-240Т) гена АСЕ находится в промоторе гена и связан с концентрацией ангиотензина II в сыворотке крови [19]. АПФ играет важную роль в регуляции артериального давления и минерального баланса крови посредством конвертации неактивной формы — ангиотензина I в ангиотензин II, который и опосредует все сердечно-сосудистые эффекты [20]. В исследовании EUROPE, включившем 8907 больных со стабильной формой ИБС, показано, что вариант rs4291 гена ACE ассоциирован с высоким артериальным давлением и, как следствие, со значительным увеличением риска развития ССО [21]. Кроме сердечно-сосудистой системы ген АСЕ экспрессируется в центральной нервной системе наряду с субстанцией Р, в основном в базальных ганглиях и в меньшей степени в гипоталамусе и других отделах мозга [22, 23]. Показано, что данный полиморфный вариант ассоциирован с развитием униполярной депрессии [24], которая является самостоятельным ФР неблагоприятного течения ИМ [25, 26].
 Различия по частотам аллелей и генотипов варианта rs4291 (А-240T) АСЕ выявлены при анализе подгрупп больных с разным годовым исходом по смертности (см. табл. 5). Носительство аллеля Т (генотипы АТ и ТТ) связано с повышенным риском смерти после ИМ (OШT=3,426). OШ для гетерозиготного генотипа АТ при сравнении с гомозиготами АА составило 9,440 и OШ для генотипа ТТ — 15,981 (OШAT vs АА=9,440 при 95% ДИ от 0,511 до 174,215; p=0,0415); OШТТ vs AA=15,981 при 95% ДИ от 0,797 до 320,398; p=0,0126).
Различия по частотам аллелей и генотипов варианта rs4291 (А-240T) АСЕ выявлены при анализе подгрупп больных с разным годовым исходом по смертности (см. табл. 5). Носительство аллеля Т (генотипы АТ и ТТ) связано с повышенным риском смерти после ИМ (OШT=3,426). OШ для гетерозиготного генотипа АТ при сравнении с гомозиготами АА составило 9,440 и OШ для генотипа ТТ — 15,981 (OШAT vs АА=9,440 при 95% ДИ от 0,511 до 174,215; p=0,0415); OШТТ vs AA=15,981 при 95% ДИ от 0,797 до 320,398; p=0,0126).
При анализе подгруппы больных с известным исходом по рецидивирующему нефатальному ИМ (n=146) проведен анализ ассоциаций с комплексной «конечной точкой» (ИМ или смерть в течение года) (см. табл. 5). Как и в случае выборки пациентов с известным годовым исходом по смертности, получены различия по частотам аллелей полиморфизма А-240Т гена АСЕ (см. табл. 5). ОШ для аллеля Т составило 2,083 (р=0,038).
Вариант rs5918 (Leu59Pro) гена тромбоцитарного гликопротеина IIIa (ITGB3) ассоциирован с повышением агрегации тромбоцитов [27], усилением связи фибриногена с GP рецепторами IIb/ IIIa [28] и уменьшением времени кровотечения [29]. Эти эффекты подтверждаются исследованиями, в которых показано, что полиморфизм rs5918 ассоциирован со снижением антиагрегантной активности ацетилсалициловой кислоты [30], риском развития ИМ вследствие коронарного тромбоза [31, 32].
В настоящем исследовании генотип по ITGB3 являлся одним из трех независимых предикторов осложнений ИМ, вошедших в модель оценки риска (см. табл. 4), однако при однофакторном анализе не был достигнут уровень статистической значимости при сравнении частот аллелей и генотипов у пациентов с разным годовым исходом (см. табл. 5).
При анализе общей выборки больных ИМ (n=165), для которой были собраны данные о статусе через год, выявлено, что генотип CG полиморфного варианта rs328 (S447X) гена липазы липопротеинов LPL связан с неблагоприятным прогнозом (см. табл. 5). Зафиксирована более высокая частота гетерозигоного генотипа в выборке с неблагоприятным исходом (37,5% против 12,3%). ОШ для этого генотипа составило 4,295 при сравнении с гомозиготами СС, второй гомозиготный генотип (GG) в выборке зафиксирован не был (OШ=4,295 при 95% ДИ от 0,949 до 19,435; p=0,0416).
Ген LPL ответственен за синтез липопротеинлипазы, которая является ключевым ферментом липолиза и регуляции метаболизма триглицеридов [33]. Липопротеинлипаза экспрессируется в основном в адипоцитах и мышечных клетках; кроме того, она связывается с эндотелием в просвете артерий и капилляров периферических тканей. Ее функция заключается в гидролизе триглицеридов липопротеинов плазмы крови до свободных жирных кислот и глицерина, а также в конвертации липопротеинов очень низкой плотности до липопротеинов низкой плотности, кроме того, липопротеинлипаза способствует связыванию липопротеинов со специфическими рецепторами [34, 35]. В многочисленных исследованиях показано, что высокий уровень триглицеридов является независимым фактором риска развития ССЗ [36, 37]. Ser447Stop (rs328) представляет собой замену С на G, которая превращает серин в положении 447 в преждевременный стоп-кодон, что приводит к формированию более короткой молекулы липопротеинлипазы. Результаты мета-анализа подтверждают, что вариант Ser447Stop ассоциирован с уровнями липидных фракций и является доказанным ФР коронарного атеросклероза [38].

При анализе подгруппы больных с разными годовыми исходами показана роль полиморфного варианта rs10811661, располагающегося вблизи генов ингибиторов циклинзависимых киназ 2А и 2B (CDKN2A и CDKN2B) в локусе 9р21, как фактора, играющего роль в развитии осложнений ИМ (см. табл. 5). Носительство частого аллеля Т, по данным литературы, является ФР развития сахарного диабета 2-го типа (OШ=1,2; р=0,0022) [39]. В настоящем исследовании показано, что редкий генотип СС обладает протективным эффектом в отношении рецидива ИМ/смерти в течение года (OШ=0,119 при 95% ДИ от 0,016 до 0,908; p=0,0158).
Выводы
Показано, что анализ небольшого количества генетических маркеров достаточен для построения модели оценки риска развития осложнений инфаркта миокарда (рецидив инфаркта миокарда/смерть в течение года), сравнимой по прогностической значимости с используемыми в настоящее время в клинической практике подходами, основанными на анализе традиционных факторов риска. При анализе 32 полиморфных генетических маркеров с доказанной ролью в патогенезе различных сердечно-сосудистых заболеваний, наилучшая модель стратификации пациентов с инфарктом миокарда по риску развития осложнений в течение 1 года включала варианты rs4291 (А-240Т) гена ACE, rs6025 (G1691A, лейденская мутация) гена F5 и rs5918 (Leu59Pro) гена IGTB3. Для подтверждения клинической ценности данная модель должна быть апробирована на независимых выборках пациентов с инфарктом миокарда. При анализе ассоциаций 32 маркеров с «конечными точками» показано, что носительство аллеля Т (генотипы АТ и ТТ) по варианту rs4291 (А-240Т) гена АСЕ и генотип СG по варианту rs328 (Ser447Stop) гена LPL являются факторами риска смерти в течение года после инфаркта миокарда.
Лейденская мутация (rs6025) гена F5 связана с высоким риском повторного нефатального инфаркта миокарда/смерти в течение года после инфаркта миокарда.
Генотип СС полиморфного варианта rs10811661, располагающегося в локусе 9р21, оказывает протективный эффект в отношении рецидива инфаркта миокарда/смерти в течение года.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований (номер проекта 13-04-021620-А).


