
Морфологическим субстратом острого коронарного синдрома (ОКС) в большинстве случаев является внутрикоронарный тромб, формирующийся в результате повреждения атеросклеротической бляшки (АСБ). Согласно результатам многочисленных исследований, выполненных в последние годы, дестабилизация АСБ в значительной степени связана с воспалительными процессами. Подтверждением этого служит обнаружение в нестабильных АСБ моноцитов, макрофагов, дендритных клеток и активированных Т-лимфоцитов, приводящих к повреждению эндотелия и внутрисосудистому тромбообразованию. Как следствие у больных с ОКС в периферической крови обнаруживаются признаки системной воспалительной реакции — повышение уровня С-реактивного белка (СРБ), интерлейкинов и других маркеров. В литературе опубликованы результаты целого ряда исследований, в которых выявлена четкая связь повышенного уровня маркеров воспаления и риска развития неблагоприятных осложнений у пациентов с ОКС [1, 2].
В последнее время внимание исследователей привлекает изучение роли иммунной системы в развитии атеросклероза. Это связано, главным образом, с тем, что иммунные процессы являются триггером воспалительных реакций. Показано, что мутации гена, ответственного за синтез белка клеточной поверхности, активирующего иммунные клетки, приводит к увеличению риска развития атеросклероза и ишемической болезни сердца (ИБС) [3]. Данные экспериментальных работ указывают на важную роль иммунной системы в атерогенезе [4].
Все большее внимание привлекает изучение участия факторов врожденного иммунитета в патогенезе ОКС: речь идет о моноцитарно-макрофагальной системе, системе комплемента, активности фагоцитоза.
Более 40 лет изучается роль активации комплемента в развитии реперфузионного повреждения миокарда у больных с ОКС. В 1970 г. J. Hill и Р. Ward опубликовали данные исследования, в котором продемонстрировано, что при развитии экспериментального инфаркта миокарда (ИМ) ингибирование комплемента приводило к уменьшению хемотаксиса и аккумуляции нейтрофилов в зоне повреждения [5]. Комплемент является одним из участников воспалительной реакции [6]. Ингибирование комплемента представлялось перспективным и обоснованным патогенетическим лечением больных с ИМ. В отдельных небольших исследованиях ингибирование компонентов комплемента приводило к уменьшению размеров ИМ [7]. Однако 3 крупных рандомизированных исследования (COMMA, COMPLY, APEX-AMI) по изучению влияния ингибитора компонента С5 комплемента пекселизумаба на течение и прогноз у больных с ОКС не выявили преимуществ этого препарата [8—10].
В последнее время внимание исследователей привлекает один из факторов активации комплемента — маннозосвязывающий лектин (mannose-binding lectin, MBL).
 MBL относится к классу Са2+-зависимых (типа С) коллектинов, которые являются рецепторами опознавания антигена в системе врожденного иммунитета. Коллектины — семейство гликопротеинов, представляющих собой поливалентные полимерные комплексы, обладающие сродством к углеводам микробов. Коллектины распознают широкий спектр инфекционных агентов. К семейству коллектинов относят MBL, компонент комплемента С1q и сурфактантные протеины легких (SP-A и SP-D). В молекуле коллектинов различают коллагеновую часть и концевой лектиновый домен. С помощью коллагеновой области они связываются с белками крови или рецепторами фагоцитов (сС1qR, кальретикулин), а с помощью лектиновых доменов — с определенными углеводами поверхности микробной клетки (грамотрицательных бактерий, микобактерий), грибов, дрожжей, некоторых паразитов и вирусов, содержащих остатки маннозы, фруктозы и N-ацетилглюкозаминогликаны. Связав микробы, коллектины способствуют их поглощению фагоцитами. Другой важной функцией коллектинов является активация системы комплемента [11].
MBL относится к классу Са2+-зависимых (типа С) коллектинов, которые являются рецепторами опознавания антигена в системе врожденного иммунитета. Коллектины — семейство гликопротеинов, представляющих собой поливалентные полимерные комплексы, обладающие сродством к углеводам микробов. Коллектины распознают широкий спектр инфекционных агентов. К семейству коллектинов относят MBL, компонент комплемента С1q и сурфактантные протеины легких (SP-A и SP-D). В молекуле коллектинов различают коллагеновую часть и концевой лектиновый домен. С помощью коллагеновой области они связываются с белками крови или рецепторами фагоцитов (сС1qR, кальретикулин), а с помощью лектиновых доменов — с определенными углеводами поверхности микробной клетки (грамотрицательных бактерий, микобактерий), грибов, дрожжей, некоторых паразитов и вирусов, содержащих остатки маннозы, фруктозы и N-ацетилглюкозаминогликаны. Связав микробы, коллектины способствуют их поглощению фагоцитами. Другой важной функцией коллектинов является активация системы комплемента [11].
MBL — белок острой фазы воспаления, синтезируемый печенью под воздействием цитокинов воспаления. Структурно и функционально MBL подобен компоненту С1q комплемента. MBL — гликопротеин, состоящий из 228 аминокислот, в котором можно выделить N-концевой домен, коллагеноподобный гликозилированный домен, образующий длинный соединительный участок, короткий шеечный домен, представленный спиралью, и С-концевой домен, взаимодействующий с углеводом на мембране микроорганизма. Три пептидные цепи, образующие базовую субъединицу, в свою очередь, составляют четвертичную структуру белка MBL — олигомер, состоящий из 26 базовых субъединиц [12]. Молекулярная масса белка составляет 400—700 кД, молекулярная масса каждой пептидной цепи — 30 кД.
В 1987 г. доказана роль MBL в антителонезависимой активации комплемента [13]. MBL образует комплекс со специфичными сериновыми протеазами (MASP-1, 2, 3), из которых только MASP-2 обладает ферментативной активностью в отношении компонентов комплемента С2 и С4.
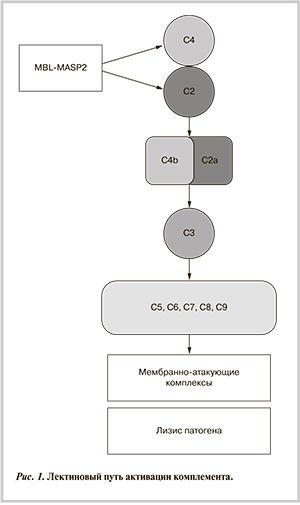
Активация лектинового пути комплемента происходит только при условии, что MBL, образовав комплекс с сериновой протеазой MASP-2, связывается со структурой, содержащей маннозу или N-ацетилглюкозоамин, например, с клеточной стенкой бактерий. Комплекс MBL—MASP-2 запускает лектиновый путь активации комплемента [14, 15]. MASP-2 расщепляет С2 и С4, образуя С3-конвертазу — С4b2a. По функциональной активности протеаза MASP-2 похожа на протеазу С1s. C1-ингибитор и α2-макроглобулин в физиологической концентрации полностью блокируют ферментативную активность комплекса MBL—MASP-2. В отсутствие ингибитора этот комплекс опсонизирует бактерии, резко ускоряя их гибель (рис. 1).
MBL образует комплекс с сериновой протеазой MASP-2. Этот комплекс расщепляет компоненты С2 и С4 комплемента, образуя С3-конвертазу (С4bC2a). Компонент С3 комплемента активирует компоненты С5, С6, С7, С8, С9. Активированный комплемент является мембраноатакующим комплексом, участвует в лизисе патогенных микроорганизмов.
Нормальный уровень MASP-2 в сыворотке составляет 170—1200 нг/мл, и MASP-2 свидетельствует о повышенном риске развития инфекционных заболеваний. Активность MASP-1 сопоставима с активностью тромбина, что позволяет предположить участие MASP-1 в системе гемостаза. MASP-3 не участвует в активации комплемента. Биологическая роль MASP-3 остается невыясненной [16].
Лектиновый путь активации комплемента является антителонезависимым, что важно для формирования быстрого иммунного ответа в случае, когда организм еще не выработал антител против возбудителя.
Помимо участия в активации системы комплемента, MBL действует как опсонин [17]. Кроме того, MBL распознает собственные поврежденные клетки и участвует в их удалении, играя важную роль в процессах апоптоза [18].
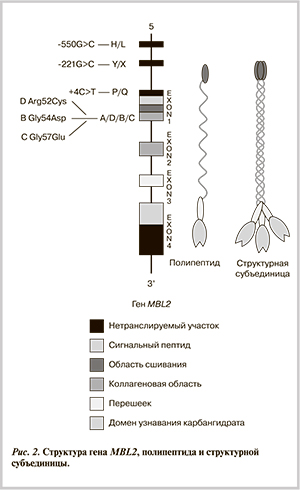
Факторы, определяющие уровень MBL в сыворотке крови. Уровень MBL в крови определяется генетическими и негенетическими факторами. R. Ezekowitz и соавт. клонировали кДНК, кодирующую MBL, в конце 80-х годов прошлого века [19]. Ген MBL локализован в области q11.2—q21 10-й хромосомы и включает 4 экзона. Экзон 1 кодирует сигнальный пептид — область, богатую цистеином и часть коллагеновой области. Экзон 2 кодирует остаток коллагеновой области, экзон 3 — шею, экзон 4 — домен узнавания карбангидрата (рис. 2).
К настоящему времени в гене MBL обнаружено 7 полиморфизмов. В промоуторной области в позиции H/L вариант, Y/X вариант, P/Q вариант в 5′-нетранслируемом регионе. В результате замены аминокислот в 1-м экзоне формируются 4 аллеля гена MBL, в кодоне 54 (глицин с аспарагиновой кислотой, аллель B), в кодоне 57 (глицин с глутаминовой кислотой, аллель C), и, наконец, в кодоне 52 (аргинин с цистеином, аллель D). Обычно наличие любого из этих аллелей обозначают «0», а нормальный аллель А называется «дикий тип». Замена глицина в 54-м и 57-м кодонах нарушает структуру α-спирали и препятствует образованию олигомера [20, 21]. Эти конформационные изменения нарушают маннозный путь активации комплемента и вызывают иммунодефицитное состояние.
Вследствие сцепления аллелей образуются 7 гаплотипов HYPA, LYQA, LYPA, LXPA, LYPB, LYQC и HYPD. Гаплотип HY ассоциируется с высоким уровнем в плазме MBL, гаплотип LY — со средним, гаплотип LX — со снижением концентрации белка MBL в плазме крови. Низкая концентрации MBL в сыворотке крови связана с наличием мутантных аллелей, локализованных в 1-м экзоне. Вариант D, в противоположность аллелям B и С, не изменяет глициновую последовательность α-спирали и связан с повышенным уровнем циркулирующего белка [22].
Обследование 7 пациентов с рецидивирующими инфекциями, представлявших 4 поколения одной семьи, показало наличие мутантных аллелей 1-го экзона гена MBL, сочетавшихся с уменьшением концентрации белка MBL в сыворотке крови и снижением возможности связывать C4b комплемента у 6 из них [23]. Это свидетельствует о том, что наследование мутантных аллелей происходит по аутосомно-доминантному типу [24].
Распространенность аллеля B среди населения северного полушария составляет 11—16%, аллеля С — не более 1%, в то время как у населения центрально-африканских стран — 0—1 и 23—29% соответственно. Ввиду очень низкой распространенности аллеля С его вклад в развитие иммунодефицитного состояния у жителей европейского континента незначителен.
Помимо генетических факторов в настоящее время изучаются негенетические факторы, определяющие уровень в сыворотке крови MBL [25]. К ним относятся возраст, пол, гормональный фон, активность иммунной системы.
Уровень MBL повышается в первые месяцы жизни и постепенно снижается к 12 годам до уровня взрослого человека [26, 27]. У недоношенных детей уровень MBL значительно ниже, чем у доношенных [28]. Как правило, уровень MBL снижается после 50 лет. У женщин он обычно выше, чем у мужчин [29].
 Нормальный уровень MBL точно не определен. Он находится в широких пределах от 10 до 5000 нг/мл. Среди представителей европеоидной расы более чем 12% популяции уровень MBL ниже 100 нг/мл. Принято считать, что уровень MBL ниже 100 нг/мл свидетельствует об иммунодефицитном состоянии. Таким образом, около 10% предположительно здорового населения страдают этим иммунодефицитом. В популяционном исследовании в Санкт-Петербурге 16% здоровых людей имели мутантный аллель MBL (по 54-му кодону 1-го экзона) и сниженный уровень MBL [30], что коррелирует с данными популяционных исследований в Западной Европе [31].
Нормальный уровень MBL точно не определен. Он находится в широких пределах от 10 до 5000 нг/мл. Среди представителей европеоидной расы более чем 12% популяции уровень MBL ниже 100 нг/мл. Принято считать, что уровень MBL ниже 100 нг/мл свидетельствует об иммунодефицитном состоянии. Таким образом, около 10% предположительно здорового населения страдают этим иммунодефицитом. В популяционном исследовании в Санкт-Петербурге 16% здоровых людей имели мутантный аллель MBL (по 54-му кодону 1-го экзона) и сниженный уровень MBL [30], что коррелирует с данными популяционных исследований в Западной Европе [31].
Уровни тиреоидных гормонов и гормона роста оказывают значительное влияние на регулирование синтеза MBL in vitro и in vivo [32, 33]. Уровень MBL отрицательно коррелирует с тиреотропным гормоном у больных с аутоиммунными тиреоидитами независимо от генотипа MBL [34]. Кроме того, уровень MBL значительно увеличивается при лечении гормоном роста [35].
Низкая активность MBL может приводить к различным клиническим проявлениям. В середине XX века выявлены случаи часто рецидивирующих инфекций у детей в результате дефицита плазменного фактора [36]. Впоследствии выяснилось, что этим фактором оказался MBL. В последнее десятилетие выполнено большое количество исследований, отражающих негативное влияние на прогноз мутаций гена MBL2, и как следствие, дефицита MBL в плазме крови больных, страдающих инфекционными заболеваниями. В основном это проявляется у лиц, адаптивный иммунитет которых либо еще не сформирован (в детской популяции), либо нарушен, как у больных с онкологической патологией после химиотерапии, ВИЧ-инфицированных.
Аллель А полиморфизма +230G/A гена и связанный с ним дефицит MBL обусловливают повышенный риск развития тяжелых форм бактериальных и вирусных инфекций у детей [37] и взрослых [38], а также инфекционных осложнений у ВИЧ-инфицированных [39-40] и онкологических больных, получающих химиотерапевтические препараты [41]. Исследование с участием 617 детей показало, что инфекционная патология у гетеро- и гомозигот по мутантным аллелям полиморфизмов структурного региона гена MBL встречалась в 2 раза чаще, чем у тех, кто не имел генетических мутаций гена MBL [42]. Гомозиготы по мутантным аллелям полиморфизмов гена MBL встречались достоверно чаще среди 337 больных пневмонией стрептококковой этиологии по сравнению с контрольной группой, включавшей 1032 пациента [43].
Преобладание гомо- или гетерозигот по вариантному аллелю выявлено у детей с менингококковой инфекцией [44], часто рецидивирующими респираторными инфекциями [45—47]. Наличие дефицита MBL связано с худшим прогнозом у детей с муковисцидозом.
Дефицит MBL является фактором риска развития диссеминированного внутрисосудистого свертывания (ДВС) у больных в ранней фазе инфекции, вызванной Staphylococcus aureus, и увеличивает риск развития синдрома ДВС при других инфекционных процессах [48].
Роль MBL при сердечно-сосудистых заболеваниях (ССЗ). В литературе имеются данные об участии активированного комплемента в атерогенезе, тромбообразовании и реперфузионном повреждении миокарда. Активация комплемента повышает риск развития ССЗ у больных с распространенным атеросклерозом. Отложение комплемента iC3b в нестабильных АСБ приводит к увеличению риска развития ОКС, по-видимому, за счет усиления воспалительной реакции и увеличения риска тромбообразования. Кроме того, оксидантный стресс, играющий ключевую роль в патогенезе ОКС, активирует комплемент через лектиновый путь в культуре клеток. Учитывая важную роль MBL в активации комплемента, можно предположить его участие в патогенезе ССЗ.
Исследования, в которых показано, что высокий уровень MBL приводит к уменьшению риска развития ИМ. S. Saevarsdottir и соавт. [49] проанализировали прогностическое значение концентрации MBL в Рейкъявикском исследовании, которое было начато в 1967 г. В общей сложности в нем приняли участие 19 382 человек. При случайной выборке образовались 2 группы случай—контроль численностью 987 и 1309 человек. В 1-ю группу вошли лица старше 70 лет, у которых развился ИМ за время наблюдения. Контрольную подгруппу составили лица старше 70 лет, у которых ИМ не развился. Больные, ранее переносившие ИМ, не включались. У больных с высоким уровнем MBL (>1000 нг/мл) риск развития ИМ был значительно ниже (отношение шансов 0,66; р<0,01). Протективный эффект MBL был наиболее выражен у больных сахарным диабетом (СД) [49]. Авторы предположили, что механизмом снижения риска развития ИМ явилось возможное участие MBL в связывании и утилизации окисленных липопротеидов низкой плотности, что приводит к снижению скорости атерогенеза.
Среди американских индейцев — носителей вариантов генотипов MBL, обусловливающих значительное снижение уровня MBL в крови, риск развития ИБС был значительно выше [50].
В небольшом пилотном исследовании, в котором приняли участие 62 больных после операции коронарного шунтирования, выявлена достоверная связь между дефицитом MBL и окклюзией венозных шунтов (p<0,01) [51].
У 91 пациента с системной красной волчанкой риск артериального тромбоза (ИМ, ишемического инсульта, тромбоза артерий нижних конечностей) был выше у гомозигот по вариантным аллелям гена MBL, имеющих сниженную концентрацию MBL [52].
У 76 больных с выраженным атеросклерозом Н. Madsen и соавт. выявили увеличение риска развития ИМ у носителей генотипа, обусловливающего низкую концентрацию MBL в сыворотке крови (p<0,01) [53].
Исследования, в которых показано, что высокий уровень MBL приводит к повышению риска развития ИМ. Вместе с тем, по данным проспективного шестилетнего исследования EPIC-Norfolk у мужчин, перенесших за 6 лет наблюдения ИМ или умерших от ИБС, исходная концентрация MBL была достоверно выше, чем у лиц, не переносивших ИМ. У женщин указанные различия не обнаружены [54]. В исследование были включены 958 больных в возрасте 64±8 лет, наблюдавшиеся в регистре EPIC-Norfolk. Контрольную группу составили 458 человек соответствующего возраста, не переносившие ИМ за время наблюдения. Не включались больные, перенесшие ИМ до начала исследования. У всех больных был определен уровень MBL и СРБ. При стратификации по квартилям MBL и СРБ наиболее высокие уровни обоих маркеров обусловливали самый высокий риск развития ИМ. Авторы связывают данный феномен с провоспалительной ролью MBL. Активируя комплемент по лектиновому пути, MBL, возможно, ускоряет процессы образования АСБ и участвует в ее дестабилизации.
Показано, что у больных СД 1-го типа риск возникновения ССЗ выше, если уровень MBL повышен. Это показано в исследовании, в котором участвовали 199 больных СД с сопутствующей нефропатией. В ряде экспериментальных работ показано, что высокий уровень MBL играет важную роль в развитии реперфузионного повреждения при ОКС с сопутствующим СД 2-го типа. Так, в экспериментальном исследовании L. La Bonte и соавт. показали, что при терапии моноклональными антителами к MBL у мышей с экспериментальным ИМ на фоне СД размер ИМ, уровень комплемента С3 и аккумуляция нейтрофилов оказались значительно ниже, чем у мышей без этой терапии [55]. Схожие данные получены в экспериментальном исследовании на мышах у М. Busche и соавт. [56].
Среди больных, перенесших каротидную эндартеркэктомию, частота рестеноза сонных артерий оказалась выше у больных с генотипом MBL, обусловливающим высокую концентрацию белка в крови. В исследовании участвовали 123 больных [57].
У больных с ревматоидным артритом высокий уровень MBL приводил к увеличению риска развития ИБС и ИМ [58].
В крупном многоцентровом проспективном генетическом исследовании среди 978 больных, перенесших операцию коронарного шунтирования, показано, что гаплотип LYQA («дикий тип», связанный с высоким уровнем MBL) — независимый предиктор послеоперационного ИМ у больных, перенесших операцию аортокоронарного шунтирования [59].
Другое направление изучения MBL у больных с ИМ — его влияние на реперфузионное повреждение. Есть предположение, что MBL может участвовать в процессах воспаления и апоптоза, являющихся ключевыми в развитии реперфузионного повреждения миокарда. Jordan J. и соавт. в экспериментальном исследовании показали, что при блокаде лектинового пути активации комплемента с помощью моноклональных антител к MBL зона реперфузионного повреждения вследствие экспериментального ИМ у мышей уменьшается. Зона реперфузионного повреждения уменьшалась за счет снижения нейтрофильной инфильтрации и экспрессии провоспалительных генов [60]. Walsh M. и соавт. выявили, что у мышей со сниженным уровнем MBL зона реперфузионного повреждения меньше, чем у мышей с нормальным уровнем MBL [61]. Авторы предположили, что в возникновении реперфузионного повреждения серьезная роль принадлежит лектиновому пути активации комплемента.
В небольшом клиническом исследовании среди 74 больных с ИМ с подъемом ST, которым проводилась первичная транслюминальная баллонная ангиопластика инфаркт-связанной артерии, уровень MBL оказался выше у больных с большим объемом повреждения миокарда (фракция выброса <35%), по сравнению с больными с фракцией выброса ≥35%. Авторы предполагают, что высокая концентрация MBL приводит к выраженной активации комплемента и увеличению реперфузионного повреждения миокарда [62].
М. Trendelenburg и соавт. [63] в исследовании с участием 890 больных с острым ИМ с подъемом ST, которым проводилась первичная ангиопластика, показали значительное повышение летальности у лиц с высоким уровнем MBL (>100 нг/мл) — 5,1 и 0,79% (р=0,02) при сравнении с больными с низким уровнем MBL (<100 нг/мл). По «комбинированной конечной точке» (смертность, кардиогенный шок, сердечная недостаточность) различий в обеих группах не выявлено. Авторы предположили, что низкий уровень MBL связан со снижением частоты развития фатальных аритмий. Происхождение данного феномена связывается со снижением реперфузионного повреждения миокарда, запускаемого с помощью MBL путем активации комплемента. Полученные данные позволяют обсуждать MBL как возможную терапевтическую мишень у больных с ОКС [63].
Таким образом, данные о влиянии MBL на течение и прогноз ИБС и ИМ неоднозначны и разнонаправлены. При этом важно отметить, что в ряде исследований не показано влияние MBL на течение ИБС. Так, среди 398 больных с доказанной ИБС полиморфизм гена MBL2, обусловливающий низкий уровень MBL, не имел отношения к прогрессированию коронарного атеросклероза [64].
Заключение
Если при инфекционных процессах имеется достаточно четкая корреляция между низким уровнем MBL и иммунодефицитом, то роль MBL в патогенезе сердечно-сосудистых заболеваний, атеросклероза, инфаркта миокарда в настоящее время остается недостаточно изученной. Полученные данные свидетельствуют о неоднозначной, комплексной роли MBL, который может в разных клинических ситуациях либо улучшать прогноз у больных, либо быть фактором риска развития осложнений. Потенциально MBL может иметь отношение ко всем основным звеньям патогенеза ишемической болезни сердца и инфаркта миокарда: воспаление, тромбообразование, апоптоз и т.д. На разных этапах атерогенеза, включая формирование и дестабилизацию атеросклеротической бляшки, тромбоз, MBL может оказывать существенное влияние. По-видимому, необходимо продолжать изучение MBL при сердечно-сосудистых заболеваниях как маркера патологии и возможной терапевтической мишени.


