
Хроническая сердечная недостаточность (ХСН) — последняя часть так называемого сердечно-сосудистого континуума, является не только сложно корректируемым состоянием, но и создает очень выраженную финансовую нагрузку на систему здравоохранения. Именно поэтому прогнозирование ее течения с целью индивидуализации лечения до сих пор остается актуальной и далекой от решения задачей. Выявлено значительное число факторов, ассоциированных с неблагоприятными исходами этого заболевания. Наиболее значимые немодифицируемые факторы неблагоприятного прогноза при ХСН — это старший возраст, пол, ишемическая этиология, ряд сопутствующих заболеваний (сахарный диабет (СД), депрессия, когнитивные расстройства, тяжелые болезни органов дыхания, хроническая болезнь почек и др.). Кроме того, значительное число факторов изменяются параллельно улучшению прогноза и могут использоваться как маркеры эффективности лечения. Так, уменьшение функционального класса (ФК), увеличение переносимости нагрузки, нормализация ритма и частоты сердечных сокращений, снижение уровня натрийуретических пептидов ассоциировано с уменьшением времени до развития неблагоприятного исхода. Описано более 100 факторов, которые могут быть ассоциированы с неблагоприятным исходом заболевания [1].
Среди многочисленных механизмов, участвующих в развитии ХСН, отдельного внимания заслуживают иммунологические процессы.
Первой работой, в которой обнаружена повышенная активность воспаления при ХСН, считается наблюдение S.K. Elster и соавт., которые в 1956 г. опубликовали сообщение, в котором описали увеличение уровня С-реактивного белка (СРБ) у 40 больных ХСН. Здесь же описано снижение у некоторых больных уровня СРБ на фоне компенсации заболевания [2]. Позже, в 1990 г., В. Levine и соавт. [3] обнаружили увеличенный уровень фактора некроза опухоли (ФНО) у больных тяжелой ХСН, осложненной кахексией.
Реакции, которые относят к разряду воспалительных, участвуют в развитии дисфункции левого желудочка, прямом негативном влиянии на сократительную способность миокарда, развитии гипертрофии миокарда, развитии фиброза миокарда, нарушениях митохондриальных процессов генерации энергии [4].
 При исследовании когорты больных из базы Finnish Acute Heart Failure Study (FINN-AKVA) на предмет ассоциации ФНО, других маркеров воспаления (интерлейкинов (ИЛ) — 6, 10), традиционных факторов (натрийуретические пептиды) с частотой смерти больных с острой декомпенсацией ХСН было обнаружено, что ФНО наряду с ИЛ-6 и мозговым натрийуретическим пептидом (BNP), возрастом и хронической почечной недостаточностью являются независимыми предикторами смерти в течение года после острой декомпенсации [5]. В то же время исследований прогностической значимости полиморфизма генов ИЛ-6 (IL-6) и ФНО (TNF) в сопоставлении с их уровнем в плазме не проводилось.
При исследовании когорты больных из базы Finnish Acute Heart Failure Study (FINN-AKVA) на предмет ассоциации ФНО, других маркеров воспаления (интерлейкинов (ИЛ) — 6, 10), традиционных факторов (натрийуретические пептиды) с частотой смерти больных с острой декомпенсацией ХСН было обнаружено, что ФНО наряду с ИЛ-6 и мозговым натрийуретическим пептидом (BNP), возрастом и хронической почечной недостаточностью являются независимыми предикторами смерти в течение года после острой декомпенсации [5]. В то же время исследований прогностической значимости полиморфизма генов ИЛ-6 (IL-6) и ФНО (TNF) в сопоставлении с их уровнем в плазме не проводилось.
Целью нашего исследования было изучение прогностической значимости определения уровня ИЛ-6, ФНО и полиморфизма их генов у больных ХСН.
Материал и методы
В исследование включен 151 больной, госпитализированный в связи с декомпенсацией ХСН. Основным критерием включения в исследование было наличие нарушения сократительной функции левого желудочка (ЛЖ). В качестве порогового было выбрано значение фракции выброса (ФВ) ЛЖ ≤40%, измеренной при трансторакальной эхокардиографии (ЭхоКГ). В исследование включались больные с ХСН различной этиологии — постинфарктный кардиосклероз, мерцательная аритмия (МА), дилатационная кардиомиопатия. В группе больных преобладали мужчины — 104 (68,9%). Средний возраст составил 64,5 года (60,9 года для мужчин и 72,8 года для женщин). Инфаркт миокарда (ИМ) ранее перенесли 111 больных (73,6%), постоянную форму МА имели 50 больных (32,8%), 132 (87,1%) имели артериальную гипертонию (АГ), 45 больных (30,2%) — СД. Среднее значение ФВ ЛЖ составило 29,6±0,57%. У 3 больных (2%) на момент стабилизации состояния ФК ХСН соответствовал I по классификации Нью-Йоркской ассоциации сердца (NYHA), у 42 (27,8%) — II, у 69 (45,7%) — III, у 37 (24,5%) — IV. Подробная клиническая характеристика больных представлена в табл. 1.
Включение больного в исследование производилось после стабилизации ХСН и принятия лечащим врачом решения о возможности выписки из стационара. У всех больных после получения письменного согласия на сбор данных и дальнейшее наблюдение производились детальный сбор анамнеза, заполнение индивидуальных регистрационных карт, трансторакальная ЭхоКГ, а также забор крови для определения уровней биомаркеров и генотипов полиморфного маркера G(–238)A гена TNF (rs361525) и полиморфного маркера G(–174)C гена IL-6 (rs1800795). Для определения аллелей и генотипов генов-кандидатов проводили выделение геномной ДНК из венозной крови обследуемых методом фенол-хлороформной экстракции. Амплификацию полиморфных участков генов проводили на амплификаторе PHC-2. Агарозные гели окрашивали бромистым этидием, полиакриламидные — нитратом серебра. Определение аллелей и генотипов изученных полиморфных маркеров генов-кандидатов проводили с помощью методик, описанных нами ранее [6].
Для анализа связи уровня N-терминального фрагмента пропептида BNP (NT-proBNP) с течением ХСН была проведена категоризация значений данного показателя на квартили: 1-й квартиль — 0—1147,6 фмоль/мл; 2-й квартиль — 1147,7—1876,7 фмоль/мл; 3-й квартиль — 1876,8—3150,6 фмоль/мл; 4-й квартиль — ≥3150,7 фмоль/мл.
Дальнейшее наблюдение проводилось посредством телефонных контактов с больным или его родственниками. Фиксировались следующие неблагоприятные клинические события: фатальный и нефатальный ИМ, фатальный и нефатальный инсульт, повторная госпитализация в связи с декомпенсацией ХСН, смерть от декомпенсации ХСН, смерть от других причин.
Статистическую обработку результатов исследования проводили с помощью стандартного статистического пакета программ SPSS 16.0. Для протяженных показателей был проведен анализ распределения и критериев его соответствия нормальному. Поскольку распределение всех изученных параметров не соответствовало нормальному, для анализа применяли непараметрические методы расчета. Для протяженных переменных рассчитывали средние величины и их ошибки (M±m). Для оценки достоверности их различия использовали непараметрические тесты Манна—Уитни и Крускала—Уоллиса. Дискретные величины сравнивали по критерию χ2 Пирсона. Правильность распределения частот генотипов определялась соответствием равновесию Харди—Вайнберга (pi2+2pipj+pj2=1) и рассчитывалась при помощи программного калькулятора Knud Christensen. Оценка независимости влияния клинических и генетических показателей на течение ХСН проводилась методом логистической регрессии. Для всех видов анализа статистически значимыми считали значения при p<0,05.
Результаты
Средний уровень NT-proBNP составил 2481,1±199,86 фмоль/мл. Среднее значение уровня ИЛ-6 составило 21,8 (±7,46) пг/мл, минимальное значение 0,71 пг/мл, максимальное 880,4 пг/мл; среднее значение ФНО-α — 10,07 пг/мл (±0,65), минимальное 10,7 пг/мл, максимальное 8,4 пг/мл. Значимых различий в уровне указанных биомаркеров в зависимости от пола обнаружено не было (табл. 2). Также не было обнаружено различий в уровнях изучавшихся показателей в зависимости от ФК ХСН по NYHA на момент выписки больного. Не выявлено какой-либо статистически значимой корреляции между уровнем ФНО-α, ИЛ-6 и уровнем NT-proBNP.
138 больных (91,4%) оказались носителями генотипа GG полиморфного маркера G(-238)A гена TNF, 13 (8,6%) — носителями генотипа AG, носителей генотипа АА выявлено не было. 54 больных (35,8%) имели генотип GG полиморфного маркера G(-174)C гена IL-6, 69 (45,7%) — генотип GC, 28 (18,5%) — генотип СС. Для полиморфного маркера G(-174)C гена IL-6 экспериментальное распределение изучаемых генотипов достоверно не отличалось от распределения, рассчитанного теоретически по закону Харди—Вайнберга; в отношении полиморфного маркера G(-238)A гена TNF проверка соответствия уравнению Харди—Вайнберга не проводилась вследствие отсутствия больных, гомозиготных по одному из аллелей.
Какой-либо связи между уровнями ИЛ-6, ФНО-α и носительством того или иного генотипа выявлено не было (см. табл. 2).
У носителей генотипа CC гена IL-6 по сравнению с генотипами GG и CG оказались значимо повышены уровни ИЛ-6 и NT-proBNP (см. табл. 2). Больных наблюдали до наступления первого неблагоприятного клинического события.
Мы не обнаружили каких-либо различий в частоте генотипов и аллелей изучавшихся генов у больных с различными вариантами клинического течения заболевания. Нами не было обнаружено связи между неблагоприятным клиническим течением ХСН и уровнями ИЛ-6 или ФНО, измеренными однократно перед выпиской больного из стационара.
Кроме уровня биомаркеров и распределения частот и генотипов изученных генов, в однофакторный анализ вошли такие факторы, как возраст, пол, АГ, МА, ишемическая болезнь сердца (ИБС), ИМ и инсульт в анамнезе, СД, ожирение, ФК ХСН по NYHA и терапия на момент выписки.
Все неблагоприятные клинические исходы. Среднее время дожития до наступления любого неблагоприятного клинического события было значимо короче у больных, являвшихся носителями аллеля A полиморфного маркера G(–238)A гена TNF (216±111,7 дней), по сравнению с носителями аллеля G (718±245,5 дней; р=0,007). Что касается клинических факторов, то неблагоприятные клинические исходы быстрее наступали у больных с инсультом в анамнезе — 336±120,2 дня против 736±192,8 дня для больных без инсультов (р=0,030); у больных с СД — 448±99,7 дня против 763±264,1 дня (р =0,029); у лиц с наиболее высоким уровнем NT-proBNP (4-й квартиль) — 356±162,8 дня против 735,8±81,73 дня для 1—3-х квартилей (р=0,012) (рис. 1). По данным однофакторного анализа, отношение шансов (ОШ) для носителей аллеля A полиморфного маркера G(–238)A гена TNF составило 3,48 (95% доверительный интервал (ДИ) 1,332–9,102; р=0,011), для больных с инсультом в анамнезе – 2,27 (95% ДИ 1,058-4,846; р =0,035), для больных с СД — 2,04 (95% ДИ 1,332-9,102; р =0,011), для больных с наиболее высоким уровнем NT-proBNP (4-й квартиль) — 2,34 (95% ДИ 1,182-4,644; р =0,015). После проведения многофакторного анализа оказалось, что только наличие аллеля A полиморфного маркера G(–238)A гена TNF и повышение уровня NT-proBNP оказались независимо связаны с развитием любого неблагоприятного клинического исхода (табл. 3) — ОШ 3,73 (95% ДИ 1,268-10,975; р =0,017) и ОШ 2,20 (95% ДИ 1,073-4,502; р =0,031) соответственно.
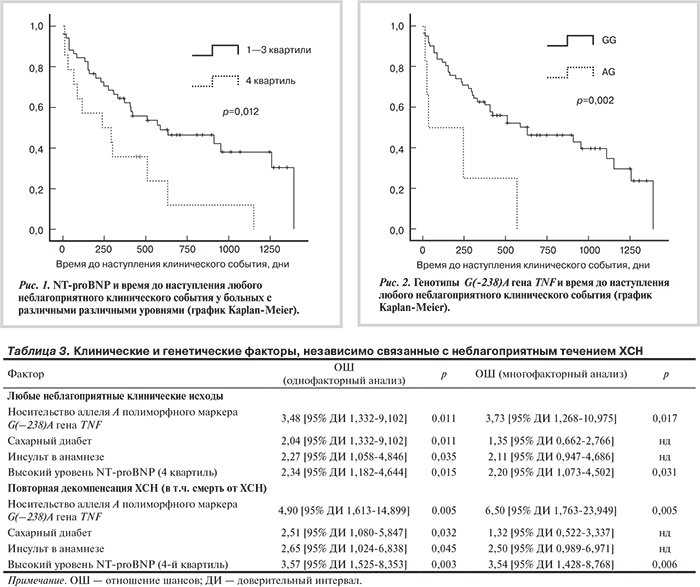
Повторная декомпенсация ХСН или смерть от ХСН. Среднее время дожития до наступления повторной декомпенсации ХСН, в том числе с летальным исходом, у больных-носителей аллеля A полиморфного маркера G(–238)A гена TNF составило 216±111,7 дня, у носителей аллеля G — 921±(77,1) день (р =0,002). У больных с инсультом в анамнезе оно составило 433±116,9 дня против 990±121,0 день для больных без инсультов (р =0,037); у больных с СД — 627±177,5 дня против 1015±227,6 дня для больных без СД (р =0,027), У больных с наиболее высоким уровнем NT-proBNP (4-й квартиль) — 456±157,2 дня против 1006±208,2 дня для 1—3-х квартилей (р=0,002). При проведении однофакторного анализа оказалось, что ОШ для носителей аллеля A полиморфного маркера G(–238)A гена TNF составило 4,90 (95% ДИ 1,613-14,899; р=0,005), для больных с инсультом в анамнезе — 2,65 [95% ДИ 1,024-6,838, р=0,045], для больных с СД — 2,51 (95% ДИ 1,080-5,847; р=0,032), для больных с наиболее высоким уровнем NT-proBNP (4-й квартиль) — 3,57 (95% ДИ 1,525-8,353; р=0,003). После проведения многофакторного анализа оказалось, что наличие аллеля A полиморфного маркера G(–238)A гена TNF и перенесенный ранее инсульт также независимо связаны с развитием повторной декомпенсации ХСН — ОШ 6,50 (95% ДИ 1,763-23,949; р=0,005) и ОШ 3,54 (95% ДИ 1,428-8,768; р=0,006) соответственно.
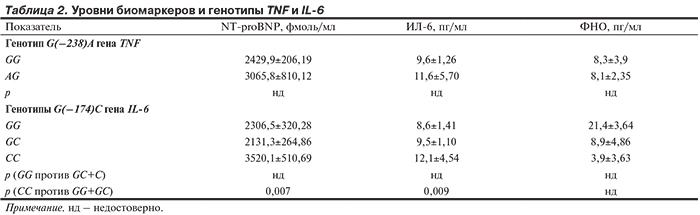
Ни один из изученных полиморфных маркеров гена IL-6 не ассоциировался с быстротой наступления неблагоприятного клинического исхода у обследованных нами больных (рис.2).
Обсуждение
ФНО — это небольшой белок, состоящий из 233 аминокислот. Вначале он синтезируется как трансмембранный белок типа II (N-конец белка находится внутри клетки), в виде гомотримера он интегрируется в мембрану, весит примерно 26 кДа. ФНО-конвертирующий фермент отщепляет экстрацеллюлярный домен, образуя растворимый ФНО массой примерно 17 кДа. ФНО синтезируется в основном в моноцитах и макрофагах. Трансмембранный ФНО также активен, выступая в двух ролях — в качестве лиганда и как рецептор [7]. ФНО является мощным провоспалительным цитокином и играет решающую роль в процессе воспаления, это активный участник реакций иммунного ответа и апоптоза. Он стимулирует экспрессию ИЛ-1, ИЛ-6, ИЛ-8. Этот цитокин влияет на функциональные свойства эндотелия, участвует в метаболизме липидов, в процессах свертывания крови, в формировании инсулинорезистентности и т.п. К настоящему времени описано гомологичных ФНО около 20 белков и 30 рецепторов, все они составляют суперсемейство ФНО [8].
Ген ФНО картирован на хромосоме 6p21.3, имеет размер 2762 последовательных нуклеотида и содержит 4 экзона. Описано значительное число полиморфных маркеров гена ФНО, значительная часть которых ассоциирована с вариабельностью экспрессии этого цитокина.
Значительное число полиморфных маркеров гена ФНО локализуется в промотерной зоне (–1031T/C, –863C/A, –857C/T, –575G/A, –376G/A, –308G/A, –244G/A, и –238G/A). Наследственный характер вариабельности уровня ФНО дает основания предполагать ее связь со структурой некодирующей части гена. Вариабельность структуры гена в этой области может приводить к различному сродству к транскрипционным факторам.
Носительство редкого аллеля A наиболее изученного промотерного полиморфного маркера G(–308)A гена TNF ассоциировано с более выраженной транскрипционной активностью, по сравнению с носительством генотипа GG [9]. Кроме того, этот полиморфизм ассоциирован с уровнем СРБ [10]. Иначе говоря, есть основания предполагать, что особенности структуры гена TNF могут определять степень воспалительного ответа.
Для оценки прогноза к настоящему моменту создано несколько шкал, описывающих прогноз для амбулаторной группы больных ХСН Heart Failure Survival Score [11], Seattle Heart Failure Model [12], и для госпитализированных больных — EVEREST Risk Model [13], шкала EFFECT [14], шкала ADHERE [15], ESCAPE Discharge Score [16], шкала —калькулятор the Barcelona Bio-Heart Failure Risk Calculator [17]. Так называемая Сиэтловская модель кроме таких факторов неблагоприятного прогноза, как ФК ХСН, ишемическая этиология, ФВ ЛЖ, систолическое артериальное давление, уровень натрия, мочевой кислоты, гемоглобина и холестерина, потребность и доза диуретика, содержит количество лимфоцитов, что косвенно может указывать на значение воспалительных реакций для течения ХСН.
При исследовании образцов миокарда, полученных во время оперативного вмешательства на сердце, было выявлено, что ФНО экспрессируется только в кардиомиоцитах больных ХСН, при отсутствии признаков сердечной недостаточности ФНО в миокарде не выявлялся [18].
Ишемия вследствие микроэмболизации может быть стимулом, вызывающим выброс ФНО из миокарда, сопровождающийся угнетением сократимости. Одновременно с этим, при экспериментальной микротромбоэмболии коронарных сосудов в сочетании с блокадой NO-синтетазы отрицательный инотропный эффект ФНО нивелируется [19].
В настоящей работе нам не удалось получить данных о связи между уровнем ФНО в крови и неблагоприятным клиническим течением ХСН. Это соответствует данным, что в целом уровни ФНО у больных ХСН не отличаются от таковых у здоровых лиц, существует значимая и тесная (r=0,6 и выше) ассоциация между уровнями BNP и концентрацией ФНО, причем эти корреляции не зависели от степени тяжести ХСН и ЭхоКГ-параметров дисфункции ЛЖ. Было обнаружено быстрое увеличение риска прогрессирования ХСН при повышении концентрации ФНО >5 пг/мл [20].
Вероятным объяснением этого факта может служить высокая степень коморбидности при ХСН, в частности, нередкое присоединение воспалительных заболеваний, таких как застойная пневмония, обострение хронического пиелонефрита, трофические поражения кожи и др. Все это приводит к высокой и быстрой вариабельности уровня ФНО и затрудняет использование уровня данного маркера в качестве самостоятельного прогностического фактора.
Нами впервые получены данные о связи между носительством аллеля A полиморфного маркера G(–238)A гена TNF и неблагоприятным прогнозом у больных с ХСН и сниженной систолической функцией ЛЖ. Эти данные косвенно подтверждаются нашим проспективным исследованием, включавшим группу больных, перенесших обострение ИБС [21]. Прогностическое значение полиморфизма TNF подтверждается и тем, что в группе отсутствовали больные-носители гомозиготного редкого аллеля, что может быть следствием их гибели до включения в исследование. Данные также соответствуют тому, что носительство аллеля A полиморфного маркера G(–308)A гена TNF ассоциировано с более выраженной транскрипционной активностью по сравнению с носительством генотипа GG [22].
Ограничением исследования является относительно небольшая выборка больных, что требует валидации полученных данных в многоцентровых проспективных исследованиях.
Таким образом, мы предполагаем, что именно генетический полиморфизм TNF, а не уровень ФНО следует использовать для предсказания развития неблагоприятных клинических исходов у больных с ХСН, наряду с другими прогностическими факторами.


