
ВВЕДЕНИЕ
Роль кардиоваскулярных нарушений в течении эндокринной патологии давно не вызывает сомнения. Начиная с 2015 г., а именно после публикации результатов проспективного исследования EMPA-REG OUTCOME [1, 2], внимание и эндокринологов, и кардиологов приковано к возможности управлять сердечно-сосудистыми исходами у пациентов с сахарным диабетом 2-го типа (СД 2) с помощью ингибиторов натрий-глюкозного котранспортера 2-го типа (иНГЛТ-2). Исследования DAPA-HF (дапаглифлозин), EMPEROR-Reduced (эмпаглифлозин), которые недавно продемонстрировали выдающуюся эффективность представителей этого класса лекарственных средств в плане снижения новых случаев госпитализаций по поводу сердечной недостаточности со сниженной фракцией выброса (ФВ), актуализируют интерес к поиску механизмов, определивших эти результаты.
Одним из эффектов иНГЛТ-2, который первоначально рассматривался исключительно как побочный, является кетонемия. Настоящий обзор призван осветить современные взгляды на роль кетоновых тел в норме и патологии с акцентом на кардио- и нейропротекцию и выходом на физиологически и эволюционно обоснованное терапевтическое использование управляемой кетонемии.
ЭНЕРГООБМЕН МИОКАРДА В НОРМЕ
Еще в 70-х гг. прошлого века было показано, что основным топливом для синтеза АТФ в митохондриях кардиомиоцитов взрослого человека в норме служат жирные кислоты (ЖК). Альтернативным, но также весьма важным энергетическим субстратом миокарда выступает глюкоза, что позволяет сердцу приспосабливаться к меняющимся условиям питания и физиологическим запросам [3]. Такая «топливная гибкость» программируется еще в пери- и постнатальном периодах. Митохондрии плода имеют сравнительно слабую окислительную активность и расходуют в основном глюкозу и лактат, тогда как ЖК используются в качестве второстепенного субстрата. В постнатальном периоде запускаются сложные транскрипционные каскады с участием в том числе ядерных рецепторов семейства PPAR, в результате чего активируются гены, связанные с окислением ЖК, и миокард становится «всеядным» [4].
Активность различных метаболических путей регулируется не только степенью экспрессии генов ключевых метаболических белков (ферментов и транспортеров), но и ферментативной и аллостерической модификацией, пространственной транслокацией, а также соотношением «продукт/субстрат» [3]. Ключевым узлом регуляции гликолиза на анаэробной стадии является фосфофруктокиназа, на аэробной – пируватдегидрогеназный комплекс (ПДГК). В окислении жирных кислот главной точкой регуляции считается карнитин-пальмитоилтрансфераза-I, ответственная за перенос ЖК в митохондрии в виде ацилкарнитина, а в качестве ключевого регулятора этого процесса выступает малонил-КоА [5]. Важную роль в управлении большинством этапов переработки энергетических субстратов миокарда играет АМФ-активируемая протеинкиназа (АМПК) [6].
60–90% ацетил-коэнзима А (ацетил-КоА) – главного топлива окислительного фосфорилирования – здоровый миокард получает от β-окисления жирных кислот, а оставшиеся 10–40% поступают от гликолиза. Исследования конца прошлого века, направленные на подавление гликолиза, предполагают, что полученная в ходе него АТФ расходуется преимущественно на перемещение Са2+ в саркоплазматический ретикулум, а также на активный ионный транспорт через цитоплазматическую мембрану. Предпринятые позже попытки подтвердить этот тезис, продемонстрировав связь гликолиза с диастолической функцией миокарда, оказались неоднозначными [7].
Активность ПДГК и соответственно аэробного гликолиза в физиологических условиях связана обратной зависимостью со скоростью окисления ЖК, что многократно показано в экспериментальных и клинических исследованиях [3].
Эффективность работы сердца в зависимости от преобладания того или иного энергетического субстрата оказывается неодинаковой. В частности, при одной и той же скорости поглощения кислорода миокардом его сократительная способность больше при утилизации глюкозы и лактата, чем при окислении ЖК. В классических экспериментах норвежского врача и физиолога Mjos O.D. у интактных собак увеличение потребления миокардом триглицеридов сопровождалось 26%-ным повышением потребления O2 без прироста механической работы левого желудочка [8]. Позже сходные результаты были получены в работах на крысах, свиньях и людях. Более того, было установлено, что подавление β-окисления ЖК в сердце крыс приводит к снижению потребления кислорода и улучшению сократимости. В дополнение к этому в 2000 г. Korvald C. et al. выявили, что подавление утилизации ЖК с помощью глюкозоинсулиновой смеси в сердце свиньи снижает в том числе и так называемое несократительное поглощение кислорода, определяемое в нижней точке кривой «давление–объем» [9].
Механизм нарушения механической эффективности и повышения расходования O2 при повышении утилизации миокардом ЖК не вполне ясен. С теоретической точки зрения окисление ЖК требует лишь немногим больше кислорода, чем необходимо для углеводов. Так, расчетное отношение АТФ/ О2 для глюкозы и лактата составляет 3,17 и 3,00, а для пальмитата и олеата – 2,80 и 2,86 соответственно [3]. В качестве причины данного феномена рассматриваются такие внутриклеточные эффекты ЖК, как митохондриальная утечка протонов и разобщение окисления и фосфорилирования [9], что снижает приведенные выше соотношения до 2,0. Тем не менее в условиях адекватной доставки кислорода окисление ЖК дает на выходе значительно больше АТФ, что и определяет их преимущество перед другими питательными субстратами.
ЭНЕРГООБМЕН МИОКАРДА ПРИ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ
При заболеваниях и состояниях, ведущих к хронической сердечной недостаточности (ХСН), энергообмен сердца существенным образом изменяется. Как показали исследования миокарда человека in vivo с использованием 31Р-МР-спектроскопии [11], общее содержание АТФ в миокарде снижается при ХСН на 40%, что сопровождается также уменьшением соотношения «фосфокреатин/АТФ» и коррелирует с выраженностью ХСН и смертностью. Такой энергодефицит частично объясняется нарушениями в цепи переноса электронов в митохондриях [12], однако существенный вклад вносит и субстратный сдвиг. В исследовании Sack M.N. et al. (1995) на материале из миокарда левого желудочка пациентов, подвергшихся пересадке сердца, продемонстрировано, что содержание ключевых ферментов окисления ЖК и их мРНК при тяжелой сердечной недостаточности снижено на 40% [13]. Доля глюкозы и лактата в энергообмене миокарда при этом соответственно возрастает. Подобную переориентацию в субстратных предпочтениях называют возвратом к «фетальному» типу метаболизма [14], с той, однако, оговоркой, что речь идет, скорее, о подавлении транскрипции ферментных систем «взрослого» типа, нежели о реактивации механизмов, оставшихся от плода.
Так называемый гликолитический сдвиг метаболизма клетки при понижении поступления кислорода известен еще с XIX в. как «эффект Пастера» и свойственен всем эукариотам. В случаях, когда такая перестройка становится постоянной или персистирующей, это сближает ее с не менее хорошо известным эффектом Варбурга, описанным для раковых клеток, что дает почву для рассуждений относительно того, не является ли гликолитическая перестройка метаболизма миокарда частью программы выживания отдельного кардиомиоцита в ущерб функции сердца как органа [15].
Интимный механизм гипоксической перестройки метаболизма долгое время оставался неизвестным, пока не были выделены и охарактеризованы особые факторы транскрипции, индуцируемые гипоксией (hypoxia-inducible factor, HIF), и прослежена их взаимосвязь с энергообменом клетки. В последние годы предметом активного изучения стали активируемые гипоксией микро-РНК (HypoxamiR), короткие некодирующие последовательности нуклеиновых кислот, которые синтезируются в ядре под влиянием HIF, проникают в цитоплазму и подавляют трансляцию определенных матричных РНК в рибосомах и митохондриях. На текущий момент уже получены данные относительно участия микро-РНК в ремоделировании сердца при различных заболеваниях, включая ХСН [16]. Подробное рассмотрение механизмов, могущих лежать в основе гипоксической перестройки энергообмена клетки, выходит за рамки нашего обзора, и содержится в специализированных публикациях [17].
Ведущие исследователи в области метаболизма миокарда признают, что изменения «субстратных предпочтений» сердечной мышцы при ХСН во многих аспектах противоречивы и не до конца понятны [18]. Принимая во внимание сказанное в предыдущем разделе о преимуществах гликолиза перед окислением ЖК в отношении расходования кислорода и сократительной способности сердца, гликолитический сдвиг метаболизма кардиомиоцитов при ХСН можно было бы считать оптимальным. Следует, однако, учитывать, что снижение митохондриальной окислительной активности касается не только ЖК, но и пирувата – промежуточного продукта гликолиза [19]. Таким образом, наряду со снижением потребления свободных ЖК происходит разобщение гликолиза и окисления глюкозы, что, в свою очередь, приводит к накоплению пирувата, лактата, ионов водорода и в конечном итоге к электролитному дисбалансу с накоплением внутриклеточного Ca2+ и Na+. А это уже непосредственно нарушает сократительную способность кардиомиоцитов. Таким образом, можно заключить, что все метаболические сдвиги в миокарде на фоне ХСН в той или иной степени дезадаптивны.
РОЛЬ КЕТОНОВ В МЕТАБОЛИЗМЕ МИОКАРДА
Кетоновые тела, а именно ацетоацетат, синтезируется в печени в ходе β-окисления ЖК из двух молекул ацетил-КоА, при этом высвобождаются две молекулы свободного КоА (рис. 1).
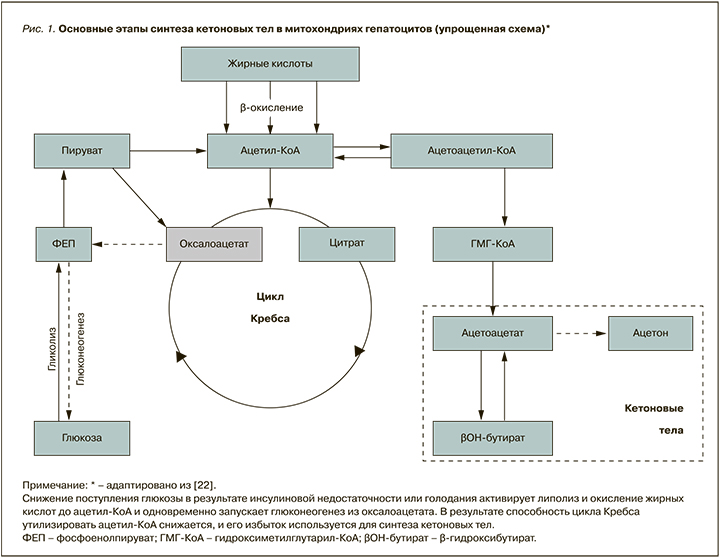
Значимость этого метаболического пути обусловлена тем, что количество свободного КоА в гепатоцитах ограничено, и при необходимости окисления избыточного количества ЖК, которая возникает, например, при голодании или дефиците инсулина, КоА быстро расходуется, а процесс окисления ЖК замедляется [20]. Не следует также забывать, что для утилизации ацетил-КоА в цикле Кребса требуется оксалоацетат, образующийся из пирувата. В условиях дефицита глюкозы не только уменьшается производство пирувата, но и активируется глюконеогенез, «оттягивающий» на себя оба метаболита. Производительность цитратного цикла при этом падает, а потребность в утилизации ацетил-КоА возрастает [21]. В этой ситуации синтез ацетоацетата решает сразу две задачи: позволяет митохондриям гепатоцитов продолжать переработку ЖК и одновременно поставлять кетоновые тела в другие ткани, где они могут вновь превращаться в ацетил-КоА и окисляться в цикле Кребса (см. рис. 1). Таким образом, кетоны – это не только способ «экспорта топлива» из печени в другие органы, главным образом в скелетные мышцы, миокард и корковое вещество почек, но и «аварийный» вариант функционирования энергообмена в условиях дефицита углеводов.
Синтез кетоновых тел возможен и в некоторых других тканях (эпителий толстой кишки, почечный эпителий, астроциты, клетки сетчатки, некоторые опухоли), но там их равновесная концентрация никогда не превышает таковую в сыворотке крови [22]. Основная транспортная форма кетоновых тел – β-гидроксибутират – поглощается тканями путем насыщаемого энергонезависимого транспорта параллельно с пируватом, при этом его проникновение внутрь клетки не зависит от инсулина. Скрупулезный анализ современных взглядов на процессы синтеза, транспорта, трансформации и утилизации кетоновых тел в различных органах и тканях содержится в недавнем специализированном обзоре [23].
С точки зрения биохимии хорошо известно, что кетоновые тела имеют метаболическое преимущество относительно глюкозы, пирувата и короткоцепочечных ЖК по причине большей внутренней энергии окисления, а в сравнении с длинноцепочечными ЖК кетонам не свойственно разобщение окисления и фосфорилирования в митохондриях. Именно этим до последнего времени объясняли их положительные эффекты, полученные во многих исследованиях на протяжении последних десятилетий при лечении различных патологических состояний, связанных с дефицитом субстратов окисления, системной и локальной инсулинорезистентностью, избытком свободных радикалов и гипоксией. К их числу относятся рефрактерная эпилепсия, онкозаболевания, врожденные дефекты ферментов гликолиза и окисления ЖК, нейродегенеративные заболевания, миопатии и даже гипогликемии при лабильном течении сахарного диабета. Традиционные взгляды на биохимическую сущность этих патологических процессов и возможности кетонов в их коррекции подробно рассматриваются в систематическом обзоре Veech R.L. [24].
Уже в середине 80-х гг. XX в. было обнаружено, что окисление кетоновых тел в миокарде тормозит окисление ЖК. Механизм угнетения β-окисления ЖК кетонами не вполне ясен, есть лишь указания на то, что изменения основных регуляторов этого процесса – концентрации малонил-КоА и соотношения «ацетил-КоА/свободный КоА» – в этом процессе не участвуют [25]. Полагают, что оно реализуется непосредственно на уровне β-окислительного каскада. Эти данные недавно подтверждены количественной митохондриальной протеомикой в экспериментальном исследовании Aubert G. et al. (2016) на мышах с гипертрофической ХСН [26]. Как и ожидалось, содержание белков, задействованных в окислении ЖК, было снижено, тогда как активность β-гидроксибутиратдегидрогеназы-1 (фермент начального этапа окисления кетонов) повышена.
Результаты ряда финских исследований конца 90-х гг. XX в. давали основания предполагать, что сердечная недостаточность сама по себе приводит к значимому повышению продукции кетоновых тел, вторичному по отношению к усилению липолиза и повышению концентрации свободных ЖК, при этом в сыворотке увеличивается содержание обоих субстратов [27]. Однако в современном проекте, использующем достижения метаболомики, напротив, показано, что содержание в сыворотке ацетоацетата и 3-гидроксибутирата у больных ХСН со сниженной ФВ существенно ниже, чем у лиц без ХСН [28], причем у больных с сохраненной ФВ уровень кетоновых тел оказался выше и не отличался от здоровых.
В исследовании Bedi K.C. Jr et al. (2016) изучались метаболомические, а точнее липидомические «слепки» сыворотки крови и биоптатов миокарда у больных с конечной стадией ХСН без диабета и болезней накопления, подвергавшиеся пересадке сердца или имплантации устройств вспомогательного кровообращения, в сравнении с пациентами без ХСН, в качестве которых выступали доноры органов [29]. В кардиомиоцитах больных ХСН было выявлено существенное снижение содержания длинноцепочечных ацилкарнитинов в сочетании с сокращением пула низших промежуточных метаболитов цикла Кребса, нормальным уровнем малонил-КоА и значительно повышенным содержанием ацетил-КоА. Содержание кетонов (β-гидроксибутирата) в сыворотке больных ХСН было выше, а в миокарде ниже, чем в контроле, в то время как уровень β-гидроксибутирил-КоА в сердечных биоптатах оказался был высоким. Это, по мнению авторов, свидетельствует о снижении захвата и утилизации ЖК в миокарде при ХСН на фоне повышенного окисления кетоновых тел, что в рамках того же исследования подтверждено активацией генов, ответственных за соответствующие ферментные системы. В качестве одного из механизмов подавления окисления ЖК может рассматриваться его субстратная регуляция избытком ацетил-КоА [3], однако дизайн исследования не предусматривал определение источника ацетил-КоА, каковым, кроме кетонов, могли также выступать углеводы и сами ЖК [29].
До последнего времени не было ясности относительно того, являются ли изменения метаболизма миокарда при ХСН адаптивными, т.е. направленными на сохранение жизнеспособности и функционального состояния, или дезадаптивными, связанными с прогрессированием основного патологического процесса. Разрешить эти вопросы попытались Horton J.L. et al. (2019) в многоэтапном экспериментальном исследовании [30]. На первом этапе с использованием генной инженерии была выведена популяция лабораторных мышей, гетерозиготных по дефициту кардиоспецифической D-β-гидроксибутиратдегидрогеназы-1 (BDH1), чтобы соответствующая часть их потомства вовсе не имела этого фермента (BDH1-/-), но оставалась жизнеспособной и фенотипически не отличалась от здоровых особей своего помета, которые использовались в качестве контроля. В опытах с перфузией изолированных мышиных сердец растворами 13С-меченых метаболитов с последующей спектроскопией ядерного магнитного резонанса (ЯМР-спектроскопией) было продемонстрировано, что миокард BDH1-/- не способен окислять D-β-гидроксибутират до ацетил-КоА.
На втором этапе того же исследования мышей подвергали 24-часовому голоданию с последующей эхокардиографией (ЭхоКГ), которая выявила значительное ухудшение функции левого желудочка и увеличение его объема в группе BDH1-/-; это подтверждает адаптивную роль кетонов в энергоснабжении миокарда в условиях физиологического пищевого стресса.
На третьем этапе у мышей обеих групп формировали хирургическую модель сердечной недостаточности (поперечное сдавление аорты плюс апикальный инфаркт) с регистрацией ЭхоКГ-показателей и транскрипционных сигнатур сердечного ремоделирования через 4 нед после операции. У мышей с дефицитом BDH1 ремоделирование и дисфункция левого желудочка оказались также более выражены, чем у исходно здоровых животных. Кроме того, часть животных контрольной группы за 1 нед до и 4 нед после операции получали кетогенную диету, и у них дилатация левого желудочка оказалась значительно менее выраженной, чем у тех, кто получал обычный корм.
В ходе четвертого этапа влияние кетонов на ремоделирование миокарда было изучено на модели дилатационной кардиомиопатии, вызванной частой кардиостимуляцией у собак, которая характеризуется хорошей воспроизводимостью и по многим параметрам близка к застойной ХСН у человека. С конца второй и до конца четвертой недели частой кардиостимуляции собаки получали β-гидроксибутират по катетеру, установленному в правый желудочек, а по окончании указанного периода выполнялась ЭхоКГ и производилось исследование скоростей субстратного окисления путем внутрикоронарного введения изотопов. Эффект от инфузии кетонов оказался разительным в отличие от контрольной группы, развитие дилатационной кардиомиопатии резко замедлялось или даже прекращалось, улучшались показатели сократимости и центральной гемодинамики, в том числе периферическое сопротивление. Окисление субстратов также существенным образом изменялось. Если у контрольных животных субстратные сдвиги в целом соответствовали описанным выше представлениям о патогенезе ХСН (подавление окисления ЖК и повышенная утилизация глюкозы и лактата), то на фоне инфузии β-гидроксибутирата утилизация ЖК существенным образом не изменялась, но снижалось поглощение и окисление глюкозы. Эти субстратные сдвиги вдобавок сопровождались увеличением поглощения кислорода.
Наконец на последнем этапе исследования изучалась эффективность окислительного фосфорилирования в изолированных митохондриях мышиных сердец, при этом было установлено значимое ее повышение под влиянием физиологических концентраций β-гидроксибутирата у здоровых мышей и отсутствие аналогичного эффекта у BDH1-/--особей. Результаты этого выдающегося исследования свидетельствуют, что по крайней мере в рамках использованных моделей повышение утилизации сердцем кетоновых тел служит адаптивной реакцией на пищевой и гемодинамический стресс. Исследователи заключают, что кетоны оптимизируют субстратное соотношение, улучшают использование O2, повышают эффективность митохондриального окислительного фосфорилирования, за счет чего обладают протективным действием на миокард.
Данные о позитивном влиянии целенаправленного введения кетонов на сердце получены и на людях. В исследовании, выполненном группой ученых из Дании, изучалось влияние длительной инфузии 3-гидроксибутирата на миокардиальный кровоток, поглощение миокардом глюкозы и пальмитиновой кислоты методом позитронно-эмиссионной томографии, совмещенной с компьютерной томографией (ПЭТ-КТ) на фоне низкодозового гиперинсулинемического эугликемического клэмпа (для подавления эндогенной продукции кетонов) у здоровых добровольцев [31]. Результатом стало двукратное уменьшение утилизации глюкозы миокардом при сохраненном темпе окисления ЖК, что сопровождалось увеличением кровенаполнения сердечной мышцы на 75%.
Как следует из приведенных работ, метаболизм кетоновых тел в миокарде сложен, при этом их позитивное влияние на энергоэффективность и производительность не вызывает сомнения.
РЕГУЛЯТОРНЫЕ ФУНКЦИИ КЕТОНОВ
По умолчанию считается, что кетоновые тела как относительно простые молекулы участвуют в регуляции метаболических процессов, изменяя равновесие биохимических реакций в качестве их субстрата или в некоторых случаях индуцируя аллостерическую модификацию ферментов. Между тем еще в 1968 г. было экспериментально обнаружено, что инфузия β-гидроксибутирата здоровым добровольцам вызывает достоверное подавление липолиза в жировой ткани без участия инсулина. Лишь спустя почти 40 лет было показано, что D-β-гидроксибутират является эндогенным лигандом рецепторов PUMA-G, через которые оказывают свое гиполипидемическое действие препараты никотиновой кислоты, причем в отличие от последней кетоны проявляют антилиполитический эффект в физиологических концентрациях.
Упомянутые рецепторы, ранее считавшиеся орфанными, сопряжены с Gi-белком цитоплазматической мембраны адипоцитов, их активация последовательно вызывает подавление аденилатциклазы, снижение уровня цАМФ и активности протеинкиназы А и быстрое ингибирование гормоночувствительной липазы, в результате чего замедляется липолиз и высвобождение свободных ЖК [32]. Концентрация кетонов сыворотки, достаточная для активации рецепторов, достигается после 2–3 дней голодания, в результате чего формируется петля обратной связи, позволяющая эффективно использовать энергию жировых депо без развития кетоацидоза. В соответствии с современной номенклатурой рецепторы никотиновой кислоты обозначаются как рецепторы гидроксикарбоновой кислоты 2-го типа (hydroxycarboxylic acid, HCA2), или G-протеин-сопряженные рецепторы 109А (GPR109A-рецепторы). Помимо адипоцитов, HCA2-рецепторы представлены на иммунных клетках (макрофаги и нейтрофилы), пигментных клетках сетчатки, эпителии толстой кишки, кератиноцитах, в микроглие и ткани молочной железы, а в минимальной пропорции и во многих других тканях, включая сердце. Наряду с липолитическим действием активация этих рецепторов вызывает глубокий противовоспалительный и иммуномодулирующий эффект, опосредованный интерлейкинами, простагландинами, фактором некроза опухоли, адипонектином и внутриклеточными модуляторами резистентности и апоптоза. Это позволяет использовать агонисты HCA2 для лечения рассеянного склероза, псориаза, нейродегенеративных заболеваний; имеются данные о возможности влиять через рецепторы этого типа на формирование атеросклеротической бляшки, субклиническое воспаление жировой ткани, опухолевые процессы в толстой кишке и молочной железе. Подробное обсуждение накопленных к настоящему моменту знаний о роли HCA2-рецепторов в этих и других процессах содержится в специальном обзоре Graff E. et al., вышедшем в 2016 г. [33].
При детальном рассмотрении регуляторные возможности кетоновых тел оказываются удивительно многообразны. Кроме HCA2, β-гидроксибутират способен взаимодействовать с рецепторами свободных ЖК 3-го типа (FFA3, или GPR41-рецепторы), которые также ассоциированы с Gi-белком. Активация и ингибирование FFA3-рецепторов задействованы в регуляции симпатического тонуса, гомеостаза глюкозы и воспалительных реакциях. Рецепторными эффектами влияние кетонов на внутриклеточные процессы не исчерпывается. В физиологических концентрациях β-гидроксибутират предположительно путем конкурентного ингибирования каталитического фрагмента снижает активность деацитилаз гистонов (HDAC) I класса – семейства ядерных ферментов, играющих важную роль в регуляции экспрессии генов посредством деацетилирования лизиновых остатков гистоновых и негистоновых белков. Вследствие подавления деацетилаз происходит гиперацетилирование гистонов, приводящее к усилению экспрессии различных генов. Описано даже β-гидроксибутирилирование гистонов, но его функциональная роль пока не ясна. К негистоновым белкам, подверженным деацетилированию HDAC, причисляют ядерный фактор NF-kB, играющий важную роль в воспалении. Наконец, имеются экспериментальные данные, указывающие на способность кетоновых тел изменять пропускную способность калиевых каналов (в частности, АТФ-чувствительных) и регулировать функцию белков-переносчиков (например, везикулярного переносчика глутамата VGLUT2). Эти и другие «сигнальные» свойства кетонов подробно обсуждаются в специализированном обзоре, опубликованном в 2017 г. [34].
Применительно к способности кетонов ингибировать HDAC вызывает интерес тот факт, что новый селективный ингибитор гистоновых деацетилаз – гивиностат – в доклинических экспериментах продемонстрировал возможность уменьшать диастолическую дисфункцию миокарда, связанную с артериальной гипертензией или возрастом [35].
Еще одна «неканоническая» функция кетоновых тел совсем недавно показана группой исследователей из США. Несколькими годами ранее они обнаружили связь субклинического воспаления жировой ткани и инсулинорезистентности с конкретным фактором врожденного иммунитета – NLRP3-инфламмасомами макрофагов и нейтрофилов, а в дальнейшем продемонстрировали возможность подавлять их сборку с помощью введения извне β-гидроксибутирата, по мнению авторов, как раз посредством уменьшения выходящего тока через калиевые каналы. Теперь же они исследовали влияние кетогенной диеты на клеточный состав жировой ткани у мышей. В итоге выяснилось, что применение кетодиеты уже в течение первой недели вызывало благоприятные изменения состава персистирующих в висцеральном жире клеток иммунной системы, а именно уменьшение числа макрофагов и увеличение количества gd-Т-клеток с протективной сигнатурой экспрессии генов [36]. Эти находки, с точки зрения авторов, приоткрывают завесу над интимными взаимосвязями иммунитета и метаболизма.
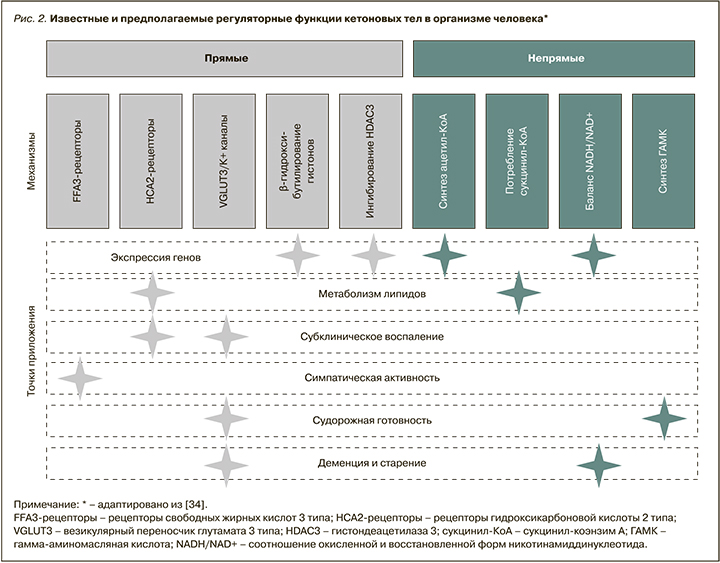
Перечисленные выше регуляторные функции кетонов суммированы на рисунке 2.
КЕТОНЫ И ГОЛОВНОЙ МОЗГ
Благоприятное влияние голодания на течение заболеваний головного мозга (конкретно – эпилепсии) известно с античности. В эпоху научного развития медицины это положение многократно подтверждено. К началу третьего тысячелетия убедительно доказан нейропротективный эффект гипокалорийного питания не только при эпилепсии, но и многих острых и хронических заболеваниях центральной нервной системы (ЦНС), таких как инсульт, паркинсонизм и болезнь Альцгеймера. Сходные результаты получены также для низкоуглеводных высокожировых диет, и, хотя оба эти подхода с очевидностью объединяет развивающаяся в ходе их применения кетонемия, роль самих кетонов в нейропротекции до последнего времени оценивалась весьма осторожно [37]. Между тем еще в конце 60-х гг. XX в. ставшие классическими эксперименты Owen O.E. и Cahill J. Jr показали, что в условиях голодания основным источником энергии для головного мозга служат кетоновые тела, вопреки и по сей день бытующему предубеждению против кетонов как эндогенных токсинов [38].
Современные исследования свидетельствуют, что значение кетонов для головного мозга не ограничивается пассивным участием в энергетическом и пластическом обмене. Прежде всего наиболее интересовавший неврологов противоэпилептический эффект кетоновых тел был экспериментально доказан in vitro и in vivo, при этом оказалось, что прямым противосудорожным действием обладают только ацетоацетат и ацетон, зато β-гидроксибутират способен снижать судорожную готовность лабораторных животных при регулярном повторном введении [39]. Нейропротекторный эффект кетогенных диет также подтвержден при непосредственном использовании кетонов на экспериментальных моделях острых и хронических повреждений головного мозга.
Наиболее популярным объяснением нейротропного действия кетоновых тел традиционно считается улучшение функции митохондрий за счет оптимизации окислительного фосфорилирования и уменьшения оксидативного стресса, а применительно к эпилепсии – модификация ГАМК-эргической передачи нервного импульса [40]. Между тем в последние годы становится все более очевидным, что описанные для других тканей регуляторные свойства кетоновых тел, обусловленные эпигенетическими, рецепторными и цитокин-опосредованными эффектами, находят свою реализацию и в ЦНС. В частности, на мышах продемонстрирована связь кетонемии с экспрессией нейронспецифических PPARg, которая регулируется упомянутыми выше гистоновыми деацетилазами [41]. А совсем недавно группа китайских исследователей на трансгенной мышиной модели болезни Альцгеймера и культуре нейронов мышиного гиппоккампа получила прямые доказательства того, что улучшение когнитивной функции на фоне лечения лабораторных животных β-гидроксибутиратом опосредовано его взаимодействием с HCA2-рецепторами [42]. В совокупности с данными, приведенными выше, это указывает на возможное наличие у кетонов нейропротекторного эффекта и потенциально раскрывает один из его механизмов.
ВОЗМОЖНОСТИ КОРРЕКЦИИ ЭНЕРГЕТИЧЕСКОГО ОБМЕНА МИОКАРДА ПРИ ПАТОЛОГИИ
Несмотря на очевидную роль нарушений метаболизма миокарда в патогенезе сердечной недостаточности любого генеза, ориентированная на коррекцию метаболизма терапия остается малоразработанной в первую очередь из-за недостаточного понимания специфики этих нарушений [18].
Исторически первым подходом к оптимизации обмена веществ в поврежденном миокарде стало использование глюкозо-инсулин-калиевой, или «поляризующей», смеси. Ее эффект заключается в улучшении доставки глюкозы и в снижении поступления жирных кислот (из-за подавления инсулином липолиза в жировой ткани) в кардиомиоциты. Частичное подавление окисления ЖК в миокарде уменьшало ишемическую дисфункцию и повреждение в экспериментальных моделях и оказывало позитивное влияние на пациентов с хронической ишемией и ХСН в клинических исследованиях. Полагают, что этот эффект реализуется благодаря пропорциональной активации окисления пирувата, меньшего накопления протонов и недоокисленных остатков жирных кислот [18].
В новую фармакологическую эпоху наиболее изученным и широко апробированным средством этого ряда является триметазидин. Считается доказанным, что он частично переключает метаболизм сердечной мышцы с окисления свободных ЖК на глюкозу за счет ингибирования митохондриальной длинноцепочечной 3-кетоацил-КоА-тиолазы. Недавний метаанализ исследований применения триметазидина при сердечной недостаточности позволил сделать вывод о его отчетливом положительном эффекте в отношении систолической функции и ремоделирования левого желудочка [43]. Следует упомянуть, что в исследовании MacInnes A. et al. (2003) влияние триметазидина на упомянутый выше фермент и в целом на окисление ЖК подтверждено не было [44]. В той же работе авторы исключили подавление этой тиолазы другим кандидатом на коррекцию метаболизма миокарда – ранолазином, все же оставив за ним способность частично снижать β-окисление свободных ЖК. Позже действие ранолазина на миокард было расшифровано как связанное с трансмембранными натриевыми каналами.
К препаратам, безусловно подавляющим окисление ЖК в миокарде, относятся ингибиторы карнитин-пальмитоил-КоА-редуктазы, блокирующие перенос ЖК из цитозоля в митохондрии, – оксфеницин, этомоксир и пергексилин. В разное время они показали эффективность у больных сердечной недостаточностью, однако первые два сняты с производства повсеместно, а последний – в большинстве стран. Причина – тяжелые побочные эффекты, обусловленные гепато-, нейро- или миотоксичностью из-за избыточного накопления липидов [45].
Предпринимались попытки усилить аэробный гликолиз за счет активации окисления пирувата. Введение извне самого пирувата оказалось эффективно только при использовании интракоронарного пути, поскольку при системной инфузии препарат активно поглощается всеми тканями организма. Утилизацию пирувата можно усилить через ингибирование ПДГ-киназы, что приводит к активации ПДК. Этим свойством обладает давно известный дихлорацетат, продемонстрировавший хороший эффект в экспериментальных моделях ХСН, но используемый в клинической практике только у больных с врожденным лактатацидозом [47] по причине все той же нейротоксичности.
Помимо регуляции распределения основных субстратов энергообмена поврежденного миокарда, можно попытаться оптимизировать утилизацию какого-либо одного из них. Эту цель преследует назначение L-карнитина или его донора – L-пропионилкарнитина. Добавочный L-карнитин усиливает транспорт ЖК в митохондрии и таким образом ускоряет их окисление в цикле Кребса, а высвобождающийся пропионил может связывать избыток свободного коэнзима А [15], однако при патологии миокарда препараты карнитина стоят лишь в ряду прочих пищевых добавок.
В целом, несмотря на определенные достижения, проблема метаболической поддержки миокарда в условиях патологии остается во многом нерешенной.
ЭНЕРГООБМЕН И ИНГИБИТОРЫ НАТРИЙ-ГЛЮКОЗНОГО КОТРАНСПОРТЕРА 2-ГО ТИПА КАК ПРЕПАРАТЫ, ИМИТИРУЮЩИЕ ГОЛОДАНИЕ
Уже на первых этапах клинического использования иНГЛТ-2 у больных СД 2 стало очевидно, что эти препараты не только выводят избыток глюкозы через почки, но и вмешиваются в энергообмен [46]. Так, в раннем исследовании Ferrannini E. et al. (2013) было установлено, что прием эмпаглифлозина, помимо закономерного снижения глюкозы плазмы и увеличения глюкозурии, сопровождается изменениями в утилизации энергетических субстратов [50]. В частности, на фоне уменьшения гликемии происходит снижение секреции инсулина и увеличение секреции глюкагона, что приводит к уменьшению потребления глюкозы и к повышению ее продукции. Снижение утилизации глюкозы в рамках исследования происходило не только путем уменьшения ее окисления, но и вследствие сокращения неокислительного расходования. Общий энергобаланс организма поддерживался за счет пропорционального повышения липолиза и утилизации жирных кислот.
Через 2 года и эндокринологическое, и кардиологическое сообщества были впечатлены результатами проспективного исследования EMPA-REG OUTCOME [48], где при добавлении 10 или 25 мг иНГЛТ-2 эмпаглифлозина к стандартной терапии СД 2 было получено беспрецедентное снижение частоты основных неблагоприятных исходов, в особенности резкое (на 38%) и раннее (начиная с первых трех месяцев лечения) снижение сердечно-сосудистой смертности, частоты госпитализаций по поводу ухудшения течения ХСН (на 35%), а также замедление прогрессирования нефропатии. Позже кардиопротективные эффекты были подтверждены и для других представителей этого класса лекарственных средств [49].
Практически сразу родилась гипотеза о ведущей роли в кардиопротективном действии иНГЛТ-2, характерной для данной группы препаратов эугликемической кетонемии. Эта идея в дальнейшем развита как гипотеза «экономного субстрата» в работах и аналитических обзорах Ferrannini E. et al. [51], которые критически проанализировали потенциальный вклад достигнутых в EMPA-REG OUTCOME метаболических и гемодинамических преимуществ (снижение гликозилированного гемоглобина, мочевой кислоты, массы тела и артериального давления) в снижение смертности и госпитализаций по причине ХСН. В сравнении с уже накопленным опытом фармакологических вмешательств, направленных на коррекцию этих показателей, эффект эмпаглифлозина оказался слишком скромным, тогда как его влияние на заболеваемость и выживаемость – значительно большим. Основываясь на собственных данных относительно влияния иНГЛТ-2 на метаболизм у больных СД 2 и здоровых людей [53], в особенности на снижение соотношения «инсулин/глюкагон», усиление липолиза и кетогенеза, а также на известных метаболических преимуществах утилизации кетоновых тел, авторы предположили, что замедление прогрессирования ХСН при СД 2 на фоне лечения эмпаглифлозином является результатом дополнительных метаболических сдвигов в миокарде под влиянием персистирующей кетонемии. По их мнению, усиление утилизации кардиомиоцитами кетоновых тел оптимизирует соотношение энергетических субстратов, расходование кислорода и продукцию макроэргических соединений, что в совокупности с благоприятными гемодинамическими сдвигами и улучшением функции почек и выражается в столь ярком кардиопротективном эффекте.
Несмотря на позитивные результаты EMPA-REG OUTCOME, даже ведущие исследователи изначально относились к ведущей роли кетонемии в снижении смертности среди его пациентов скептически. В частности, высказывалось суждение, что повышение концентрации β-гидроксибутирата в сыворотке на фоне лечения иНГЛТ-2 может быть следствием не увеличения его продукции в печени, а уменьшения общего клиренса предположительно за счет снижения потребления кетонов тканями, в том числе миокардом. В показательном клиническом эксперименте исследовательской группы Ferrannini E. было убедительно продемонстрировано, что кетонемия на фоне применения иНГЛТ-2 связана именно с гиперпродукцией β-гидроксибутирата, а не уменьшением его клиренса [54].
В качестве еще одного аспекта положительного влияния эмпаглифлозина на выживаемость пациентов обсуждаемого исследования рассматривается зарегистрированное у них небольшое (в пределах 5%) увеличение гематокрита, которое может улучшать доставку кислорода к тканям [1, 55]. Первоначально предполагалось, что оно обусловлено диуретическим эффектом иНГЛТ-2. Однако двумя годами раньше при изучении гемодинамических эффектов дапаглифлозина было обнаружено, что под действием препарата увеличивался не только гематокрит, но и общая масса эритроцитов, чему предшествовало повышение концентрации эритропоэтина и числа ретикулоцитов. Увеличение продукции эритропоэтина почками зафиксировано и в упомянутом выше исследовании с применением эмпаглифлозина [54].
Как отмечают многие исследователи, изучающие эффекты иНГЛТ-2, уже имеющиеся доказательства позитивных эффектов этой группы препаратов, а также известные метаболические и регуляторные возможности кетоновых тел в оптимизации энергетических процессов в организме, в частности в миокарде, требуют дополнительных исследований, которые позволят не только предположить, но и увидеть их непосредственную взаимосвязь [51, 52]. Единичные сообщения о таких исследованиях уже появляются. Так, в совсем недавно опубликованном исследовании Byrne N.J. et al. (2020) выявлено замедление прогрессирования экспериментальных моделей сердечной недостаточности у мышей на фоне лечения эмпаглифлозином в дозе 10 мг/кг; при этом наблюдалось снижение активности мииокардиальных инфламмасом NLRP3 [56]. Примечательно, что работа выполнена на животных без диабета, и наблюдавшиеся кардиопротективные эффекты препарата не сопровождались повышением уровня кетоновых тел. Между тем двумя годами ранее у мышей с диабетом, но без сердечной недостаточности было обнаружено положительное влияние того же представителя иНГЛТ-2 на производительность и энергообмен миокарда, оцениваемый по соотношению «фосфокреатин/АТФ» при магнитно-резонансной спектроскопии, которое имело прямую корреляцию с концентрацией β-гидроксибутирата [57].
Однако наиболее убедительным подтверждением экспериментальных данных стали результаты недавно представленных исследований DAPA-HF и EMPEROR-Reduсed, в которых дапаглифлозин (в дозе 10 мг) и эмпаглифлозин (10 мг) у больных ХСН со сниженной ФВ безотносительно статуса СД показали значимое влияние на снижение госпитализаций по поводу сердечной недостаточности. Но подлинным открытием стали неожиданные результаты исследования EMPEROR-Preserved , где эмпаглифлозин 10 мг впервые среди прочих кардиотропных препаратов продемонстрировал подобные позитивные эффекты в отношении пациентов с ХСН с сохраненной ФВ.
Многие вопросы, поставленные неожиданными результатами EMPA-REG OUTCOME, DAPA -HF, EMPEROR-Reduсed, EMPEROR-Preserved, остаются открытыми. К их числу можно отнести, например, зависимость функционального состояния сердца от дозы и экспозиции как кетонов, так и иНГЛТ-2, влияние на реализацию их эффектов состояний, сопутствующих диабету и ХСН, а также применяемых параллельно препаратов, в особенности инсулина и его агонистов. Дальнейшее изучение этих проблем позволит расширить представления о патогенезе сердечной недостаточности, ее взаимосвязях с нарушениями обмена веществ, вырабатывать новые подходы к их профилактике и лечению.
КЕТОЗ И КЕТОАЦИДОЗ
Содержание кетоновых тел в крови человека и лабораторных животных хорошо изучено применительно к различным физиологическим и патологическим состояниям. Уровни суммарных кетонов (β-гидроксибутират + ацетоацетат), которые можно выявить в человеческой сыворотке, приведены в таблице.
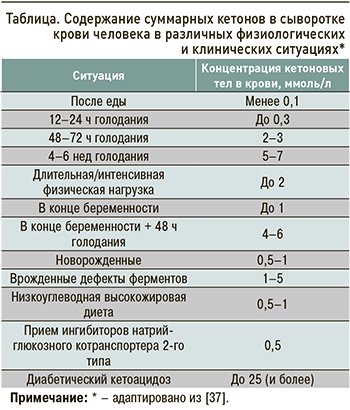
Соотношение «β-гидроксибутират/ацетоацетат» у здорового человека после еды колеблется от 1 до 2, по мере нарастания кетоза оно увеличивается до 3–10. За верхний предел нормы для кетонемии обычно принимают 0,5 ммоль/л; при 1,0 ммоль/л и более говорят о гиперкетонемии или кетозе; при 3 и более ммоль/л может развиться кетоацидоз.
Физиологическая и патологическая гиперкетонемия различаются не только количественно. Голодание у здорового взрослого человека не вызывает опасных метаболических расстройств на протяжении до нескольких недель, так как значительная часть кетонов утилизируется мышцами и ЦНС, покрывая до 80% энергозатрат головного мозга [38]. При декомпенсированном СД головной мозг человека, будучи большим и при этом инсулинонезависимым органом, продолжает утилизировать изобилующую в крови глюкозу, в то время как печень наращивает синтез кетонов. В результате кетонемия и ацидоз неуклонно прогрессируют [40].
Ацетоацетат в противовес β-гидроксибутирату обладает провоспалительными свойствами [58], что по мере перехода кетоза в кетоацидоз может нивелировать потенциальные позитивные свойства умеренной кетонемии.
Еще одно существенное отличие физиологической кетонемии от диабетического кетоза – динамика уровня кетонов в течение суток. При кетозе на фоне декомпенсации СД график концентрации кетоновых тел в крови имеет линейный характер, т.е. плавно нарастает без лечения и убывает на фоне инсулинотерапии. Физиологическая кетонемия определяется длительностью голодания и быстро купируется приемом углеводов, рецидивируя, если дефицит углеводов возобновляется [38].
Большинство упомянутых выше работ, демонстрирующих позитивные эффекты кетонов (преимущественно β-гидроксибутирата) на различные аспекты функционирования различных органов и тканей, оперирует концентрациями этого метаболита в пределах 0,5–2,0 ммоль/л, которые без большой натяжки можно считать физиологическими.
Кетонемия на фоне применения иНГЛТ-2 у больных СД 2 заметно превышает исходный уровень, при этом ее выраженность поддается прогнозированию, исходя из стартовых концентраций глюкозы, свободных ЖК и инсулина в плазме [59].
Динамика кетонемии на фоне разового приема и 4-недельного лечения эмпаглифлозином детально изучена группой исследователей из Японии [60]. Они, в частности, сравнивали суточный профиль суммарных кетонов крови в первые и 28-е сутки приема эмпаглифлозина в дозе 10, 25 мг или плацебо у пациентов с СД 2 на фоне стандартной диеты. Результаты убедительно показали, что эмпаглифлозин вызывает дозозависимую кетонемию с максимумом после ночного голодания, близким к 0,5 ммоль/л (по среднему значению), причем отдельные индивидуальные значения достигали без малого 1,45 ммоль/л. Как видно из графиков, каждый прием пищи, содержащей углеводы, как и при физиологическом кетозе голодания, снижал уровень кетонов в крови испытуемых до нормы (рис. 3).
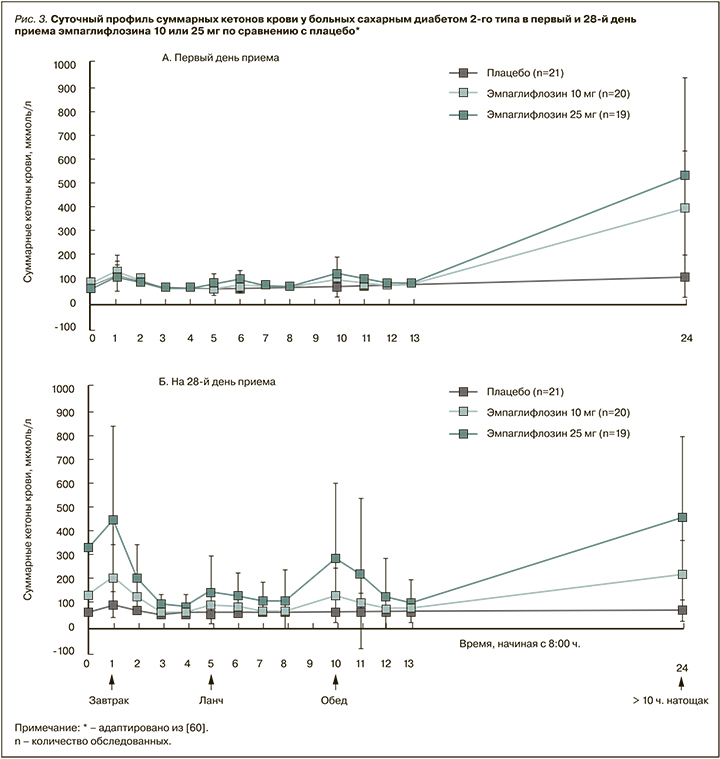
Таким образом, и по абсолютному уровню кетонов крови, и по его динамическому временному паттерну ассоциированная с иНГЛТ-2 кетонемия близка к физиологической.
ЗАКЛЮЧЕНИЕ: ОЖИРЕНИЕ, ЭВОЛЮЦИЯ И ЭНЕРГООБМЕН
Изучение кетонемии через энергетику голодания логически выводит исследователей на патофизиологию ожирения, которое посредством инсулинорезистентности связано с основными неинфекционными пандемиями – СД, ишемической болезнью сердца и онкопатологией. В пилотном исследовании Weyer C. et al. (2001) при сравнении энергообмена у 7 европейцев и 7 мужчин североамериканского индейского племени Пима было впервые продемонстрировано наличие двух его фенотипов – «метаболически экономного» и «метаболически расточительного» [61]. Всех испытуемых последовательно подвергали двухдневному голоданию и перееданию. Особенностью «расточительного» фенотипа был больший расход энергии в ответ на переедание в сочетании с меньшим его снижением при голодании, тогда как у представителей «экономного» фенотипа все выглядело наоборот.
Эти результаты позже были воспроизведены в более современных работах на представителях разных этнических групп, при этом было показано, что «экономному» фенотипу при проспективном наблюдении свойственно легче набирать массу тела при обычном, неконтролируемом питании и труднее терять ее на фоне гипокалорийной диеты [62]. Хотя существует большой массив данных относительно регуляции массы тела и пищевого поведения, конкретные энергочувствительные механизмы, связывающие расход энергии и массу тела, пока не определены [63].
Несмотря на отсутствие прямых доказательств, есть множество оснований полагать, что «экономный» фенотип энергообмена обусловлен соответствующим генотипом, который, вероятно, подвергся жесткому естественному отбору в верхнем Палеолите [64].
Первым кандидатом на «экономный» ген, а точнее «экономную» мутацию, стала утрата человеком и крупными приматами фермента уриказы, предположительно случившаяся гораздо раньше, в эпоху среднего миоцена. Это, по-видимому, дало нашим общим предкам эволюционное преимущество в виде повышенной способности аккумулировать жиры из растительной фруктозы [65].
Другие генетические детерминатны «экономного» генотипа до настоящего времени не определены, зато интуитивно понятны факторы, под давлением которых произошел их отбор. На протяжении 99,5% истории рода Homo, вплоть до широкого распространения земледелия и скотоводства, предкам современного человека приходилось выживать в условиях непредсказуемой доступности пищи, при этом еще и небогатой углеводами, будучи вынужденными периодически испытывать интенсивные физические нагрузки для ее добычи и защиты от опасностей [64]. Периоды недоедания и голода чередовались с относительно короткими эпизодами изобилия, периоды покоя – с физическим и эмоциональным стрессом. Это требовало от организма той самой метаболической гибкости, т.е. способности быстро усваивать, запасать и мобилизовать разнообразные источники энергии для мышечной и мозговой активности, функционирования жизненно важных органов и иммунной системы [66].
Слом этого древнего стереотипа на современном этапе истории человечества признается одной из ведущих причин пандемии ожирения и связанных с ним так называемых болезней цивилизации, в основе которых лежит инсулинорезистентность. Решение этой проблемы настолько же очевидно, насколько и трудно выполнимо, поскольку соответствующей эволюционно сформированной мотивации самостоятельно ограничивать себя в пище и подвергать себя «бесцельным» нагрузкам у большинства людей нет. Альтернативы физическим нагрузкам для нормального энергообмена, по сути, не существует, тогда как пищевые ограничения лежат в основе самых разнообразных диет, из которых наиболее физиологичными представляются схемы с ограничением времени приема пищи и периодическими разгрузочными днями [67]. Модулируемое ими динамическое переключение энергобмена с потребления на расходование и с гликогенолиза на липолиз возвращает утрачиваемую организмом современного человека метаболическую гибкость. Маркером и как минимум отчасти медиатором этой гибкости является периодическая физиологическая кетонемия, которая выступает самым заметным биохимическим феноменом гипокалорийного питания или голодания. Последнее в схеме прерывистого голодания во многих экспериментальных и клинических исследованиях неоднократно демонстрировало позитивное влияние на снижение частоты сердечно-сосудистых событий, новых случаев СД 2, онкологических заболеваний.
У пациентов с уже развившимся СД 2 в дополнение к диете и физическим упражнениям альтернативную возможность безопасно смоделировать периодическую кетонемию дает использование иНГЛТ-2, которые становятся в этом контексте препаратами, «имитирующими» голодание. Это открывает новые перспективы восстановления метаболической гибкости, а вместе с нею улучшения функционального состояния и долговечности как миокарда, так и других органов-мишеней.


