ВВЕДЕНИЕ
Сахарный диабет (СД) – группа метаболических (обменных) заболеваний, характеризующихся хронической гипергликемией, которая является результатом нарушения секреции инсулина, действия инсулина или обоих этих факторов [1]. СД 1-го и 2-го типа наиболее распространены среди всех форм СД в мире и относятся к мультифакториальным заболеваниям, причиной развития которых является совокупность наследственных и средовых факторов. Редкие формы диабета, развивающиеся вследствие дефекта в одном гене, составляют менее 5% случаев от всех форм СД [2]. Менделевские подтипы этих форм СД включают диабет взрослого типа у молодых, или MODY (Maturity-Onset Diabetes of the Young), неонатальный диабет, а также синдромальные формы. Трудность их диагностики состоит в том, что многие из этих подтипов имеют клинические признаки, сходные с СД 1-го и 2-го типа [3]. Пациентам с моногенными формами СД требуется персонализированный подход к подбору оптимальной терапии [4].
Верифицировать у пациента моногенную форму СД можно только при проведении молекулярно-генетического исследования. В случае его отсутствия до 80% случаев моногенного диабета неверно диагностируются или остаются нераспознанными [5]. Самой известной и хорошо изученной формой моногенного диабета является MODY. Эта гетерогенная группа заболеваний с аутосомно-доминантным типом наследования обусловлена структурными нарушениями в генах, приводящими к дисфункции бета-клеток поджелудочной железы. В настоящее время известно 14 типов MODY, которые классифицируются по мутациям генов, определяющих клинический фенотип: HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, KCNJ11, ABCC8 и APPL1 [6]. 70% случаев MODY ассоциированы с мутациями в генах GCK и HNF1A [7].
У пациентов с MODY, обусловленным носительством мутаций в гене GCK, умеренная гликемия натощак проявляется уже с рождения, обычно протекает бессимптомно и чаще всего не требует назначения сахароснижаюшей терапии. Пациенты с MODY-GCK могут корректировать уровень гликемии диетой и умеренными физическими нагрузками [8].
Гипергликемия при MODY-HNF1A диагностируется в возрасте от 21 до 26 лет, семейный анамнез у носителей, как правило, отягощен по СД и сердечно-сосудистым заболеваниям в нескольких поколениях. Натощак у таких пациентов наблюдается нормогликемия, но при выполнении орального глюкозотолерантного теста фиксируется прирост гликемии. Также для MODY-HNF1A характерна глюкозурия. Заболевание при этом носит прогрессирующий характер с высоким риском развития макрососудистых осложнений. При лечении у пациентов сохраняется высокая чувствительность к препаратам сульфонилмочевины [9]. Ген HNF1A расположен на длинном плече 12-й хромосомы (12q24.31) и состоит из 10 экзонов (https://www.uniprot.org). Белок, кодируемый этим геном, состоит из 631 аминокислотного остатка, его структура консервативна у позвоночных животных (при сравнении человека и крысы идентичность аминокислотного состава составляет 97%) [10]. В своей структуре белок HNF1A содержит N-концевой димерезационный домен (первые 32 аминокислоты), ДНК-связываюший высококонсервативный домен (91–279 аминокислот), внутри которого находится неконсервативный участок (со 182 по 200 аминокислоту), разделяющий специфический POUS гомеодомен (91–181 аминокислоты) и POUH-гомеодомен (203–279 аминокислоты), и С-концевой трансактивационный домен (282–631 аминокислот) [11, 12]. Альтернативный сплайсинг обеспечивает образование трех изоформ белка, которые различаются аминокислотным составом и уровнями экспрессии [13]. Исследования на мышиных моделях показали, что гетерозиготные нокауты по гену HNF1A имеют нормальный фенотип, тогда как у человека наличие гетерозиготного варианта приводит к развитию СД [14, 15]. Основной транскрипт кодируется на участке с 1-го по 10-й экзон и экспрессируется в период эмбрионального развития в поджелудочной железе, а в зрелом возрасте в тканях почек и печени. Более короткие изоформы, кодируемые экзонами 1–7 и 1–6, выявлены преимущественно в зрелых тканях поджелудочной железы [16]. Известно более 500 различных вариантов в гене HNF1A, которые приводят к развитию СД. Описаны как однонуклеотидные замены в различных его частях, так и структурные перестройки гена HNF1A (http://www.hgmd.cf.ac.uk/ac/search.php). Показана высокая степень пенетрантности у носителей гетерозиготных мутаций гена HNF1A; к 25 годам развитие диабета наблюдается у 63%, к 35 годам – у 78 %, а к 55 годам – у 95,5% из них [17]. Диагноз СД 2-го типа с ранним началом нередко является ошибочным у носителей мутаций в этом гене, так как раннее начало диабета (25–45 лет), наличие семейной истории и отсутствие ожирения не относятся к специфическим критериям и не позволяют проводить дифференциальный диагноз [18].
Целью данной работы было показать возможности молекулярно-генетического исследования для MODY-HNF1A и СД 2-го типа с ранним началом на примере клинического случая диабета у пробанда с отягощенным семейным анамнезом.
МАТЕРИАЛ И МЕТОДЫ
Протокол исследования был одобрен этическим комитетом НИИ терапии и профилактической медицины – филиала Института цитологии и генетики Сибирского отделения РАН (№ 7 от 22.06.2008). Письменное информированное согласие на обследование и участие в исследовании было получено от каждого пациента.
Выполнен забор образцов венозной крови у пробанда и ее матери для молекулярно-генетического исследования. Геномную ДНК выделяли из лейкоцитов венозной крови методом фенол-хлороформной экстракции [19].
Проведено таргетное высокопроизводительное секвенирование. В таргетную панель были включены кодирующие участки и прилегающие сайты сплайсинга MODY-ассоциированных генов, в том числе гена HNF1a. Для приготовления библиотек использовали набор KAPA HyperPlus Kit (Roche, Швейцария). Качество анализируемой ДНК и подготовленных библиотек оценивали с помощью системы капиллярного электрофореза Agilent 2100 Bioanalyzer (Agilent Technologies Inc., США). Анализ полностью подготовленной библиотеки проводился на платформе Illumina MiSeq (Illumina, США). Полученные данные выравнивали на референсный геном человека (GRCh37) с использованием программного обеспечения Burrow–Wheeler Alignment tool (BWA v.0.7.17) (http://bio-bwa.sourceforge.net). Последующий биоинформационный анализ включал удаление ПЦР-дубликатов, поиск однонуклеотидных вариантов с помощью Genome Analysis Toolkit v.3.3 (https://gatk.broadinstitute.org/hc/en-us) и их аннотацию с использованием программы ANNOVAR (https://annovar.openbioinformatics.org/en/latest). Также были использованы данные баз gnomAD (https://gnomad.broadinstitute.org), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), HGMD (база данных мутаций генов человека, http://www.hgmd.cf.ac.uk/ac/index.php) и учтены литературные данные. Селекцию редких и новых вариантов проводили в MODY-ассоциированных генах (HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, CEL, PAX4, INS, BLK, KCNJ11, ABCC8 и APPL1). Ранее нами было опубликовано подробное описание проведения биоинформационного анализа [20].
Обнаруженная замена rs137853240 у пробанда и ее матери была подтверждена секвенированием по Сэнгеру фрагмента ДНК, содержащего экзон 4 гена HNF1a с использованием прямого и обратного праймеров: 5’-GTCACACAGCAGACCTGGCA-3’, 5’-GGCATGAATGGAATGGAACC-3’. Дизайн олигонуклеотидов для исследованного варианта был выполнен в программе Primer-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast). Секвенирующая реакция была выполнена на приборе ABI 3500 (Thermo Fisher Scientific, США) с помощью набора BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, США). Последовательности были проанализированы в программе Vector NTI® Advance (Thermo Fisher Scientific, США). Версия hg19 человеческого генома служила референсной последовательностью для выравнивания.
На втором этапе исследования выполнен анализ распространенности аллелей и генотипов rs137853240 в выборках из популяции европеоидного населения Западной Сибири (376 человек, средний возраст 55,9±6,8 лет, доля мужчин 45,2%) и этнических групп, включающих чукчей (224 человека), коряков (56 человек) и эскимосов Канады (20 человек), а также в клинической группе пациентов с СД 2-го типа (169 человек, средний возраст 59,0±6,7 лет, доля мужчин 45%, уровень глюкозы больше 11,1 ммоль/л), обследованных в рамках проекта HAPIEE (Новосибирск) [21] с использованием технологии TaqMan. Амплификацию геномной ДНК, которая содержала изучаемый вариант, проводили методом полимеразной цепной реакции в режиме реального времени (ПЦР-РВ) на приборе StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, США). Подбор фланкирующих олигонуклеотидов и TaqMan-зондов осуществляли с помощью программы Vector NTI Advance 11.0 (Thermo Fisher Scientific, США). Были подобраны следующие фланкирующие олигонуклеотиды и комплементарные зонды, меченные красителями FAM, HEX и гасители флуоресценции BHQ1и BHQ2 соответственно: rs137853240-F-5’-CTGCCCGCTCACAGCTC- 3’, rs137853240-R-5’-CTGCCCGCTCACAGCTC-3’, rs137853240-5’-[FAM] TCCCCCAGTAAGGTCCACGGTAAGT [BHQ1]-3’, rs137853240-5’-[HEX] TCCCCCAGTAAGGTCCACAGTAAGT [BHQ2]-3’. Реакцию ПЦР-РВ проводили в объеме 25 мкл с использованием 15 нг ДНК, по 100 нМ прямого и обратного фланкирующих праймеров, 50 нМ каждого из двух зондов и мастер микса БиоМастер HS-qPCR Hi-ROX (2×) (Биолабмикс, Россия) согласно протоколу производителя. Режим амплификации начинался со 120 с при 60 °C, затем 36 циклов: 95 °C – 30 с, 66 °C – 30 с, 72 °C – 30 с. Завершалась программа амплификации при 600 °C в течение 120 с.
РЕЗУЛЬТАТЫ
Клинический фенотип MODY был выявлен у пробанда и ее матери при обследовании у эндокринолога [22]. Пациентка 26 лет обратилась к эндокринологу, в возрасте 15 лет у нее было диагностировано нарушение толерантности к глюкозе. В 16 лет появились жалобы на общую слабость, уровень глюкозы плазмы натощак составлял 9,1 ммоль/л, антитела к глутаматдекарбоксилазе были отрицательными, выявлена глюкозурия (до 3 г/л в утренней порции мочи), НbА1с – 8,0%. У пациентки определяется отягощенный наследственный анамнез по гипергликемии.
У матери пробанда был диагностирован СД 2-го типа в возрасте 39 лет (в настоящий момент ее возраст 53 года, индекс массы тела 38,3 кг/м2). Принимает пероральную сахароснижающую терапию (метформин, гликлазид и эмпаглифлозин). Определяется диабетическая дистальная полинейропатия, ожирение 2-й степени, артериальная гипертензия, неалкогольная жировая болезнь печени.
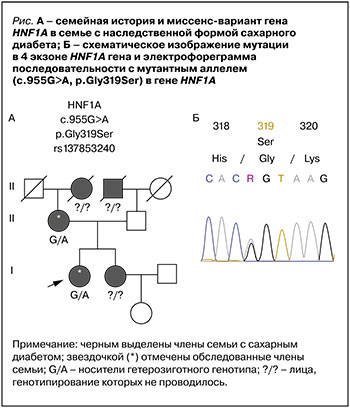
У бабушки пробанда по линии матери СД был впервые выявлен в возрасте 58 лет, для лечения применялся метформин. У дедушки по линии отца в возрасте 60 лет был диагностирован сахарный диабет 2-го типа, но у отца пробанда на момент обследования наблюдалась нормогликемия (рис., фрагмент А). При молекулярно-генетическом исследовании у пробанда и ее матери была выявлена гетерозиготная миссенс-замена c.955G>A, p.Gly319Ser (rs137853240) экзона 4 гена HNF1А (рис., фрагмент Б). Выявленный вариант описан в базе данных ClinVar, упоминается в базе данных gnomAD и описан в литературе [23] в популяции Оджи-кри. Эта этническая группа является изолированной популяцией канадских эскимосов в заповеднике Сэнди-Лейк в Северном Онтарио [24], поэтому в дальнейший анализ популяционной распространенности редкого аллеля мы включили этнические группы коряков, чукчей и эскимосов Канады.
При оценке распространенности вариантов по rs137853240 мы не обнаружили носителей c.955G>A, p.Gly319Ser (rs137853240) в выборках из популяции европеоидного населения Западной Сибири и этнических групп чукчей, коряков и эскимосов Канады, а также в клинической группе пациентов с СД 2-го типа.
Полученные нами результаты и данные предыдущих исследований позволяют предположить, что вариант c.955G>A, p.Gly319Ser (rs137853240) экзона 4 гена HNF1А служит причиной развития MODY-HNF1A у пробанда и ее матери.
ОБСУЖДЕНИЕ
Представленный случай демонстрирует особенности течения СД, который связан с мутацией в гене HNF1A, кодирующим ядерный фактор гепатоцитов-1. Как известно, у пациентов молодого возраста с нарушением углеводного обмена может быть верифицирован СД 1-го типа, СД 2-го типа или более редкие моногенные формы диабета. Такие характеристики исследованного пациента, как начало СД в молодом возрасте, сохраненная функция бета-клеток при стаже заболевания более 10 лет (уровень С-пептида находился в пределах референсных значений и составлял 469 н/мл), отсутствие ожирения, выявление дислипидемии в молодом возрасте (триглицериды – 3,1 ммоль/л, общий холестерин – 7 ммоль/л), а также отягощенный наследственный анамнез по гипергликемии, позволили предположить наличие MODY-HNF1A. Молекулярно-генетическое исследование выявило мутацию Gly319Ser в гене HNF1A у двух членов семьи с СД. Ядерный фактор гепатоцитов-1 гомеобокс А принадлежит к группе транскрипицонных факторов, которые играют важную роль в формировании и дифференцировке различных органов и тканей (печень, почки, поджелудочная железа) посредством регуляции экспрессии тканеспецифических генов в период эмбрионального развития и в течение жизни [25].
Обнаруженный нами вариант c.955G>A, p.Gly319Ser был впервые выявлен у коренного населения Канады с СД 2-го типа, в популяции Оджи-Кри [23] и не был найден ни у одной другой исследованной ранее этнической группы [23]. Также он не был обнаружен нами в популяционной выборке г. Новосибирска, в выборке пациентов с СД, в популяционных выборках коряков, чукчей и канадских эскимосов. Носительство варианта Gly319Ser в популяции Оджи-Кри коррелировало с предрасположенностью к СД 2-го типа: оно наблюдалось у 40% пациентов с этой формой заболевания [23]. В популяции Оджи-кри на фоне увеличения потребления пищевых жиров и снижения физической активности произошло многократное увеличение частоты случаев развития СД 2-го типа. В ходе поиска генетического фактора, предрасполагающего к заболеванию, идентифицирован вариант Gly319Ser гена HNF1A. Частота аллеля Ser319 составляла 0,190 (21/110) у лиц с диабетом и 0,063 (22/350) у лиц без диабета [26]. Высокая частота варианта в популяции может быть обусловлена эффектом основателя – локальным повышением частоты редкого аллеля в результате сегрегации ограниченной группы индивидуумов. Среди лиц с диагнозом СД 2-го типа носители Gly319Ser, по сравнению с остальными пациентам, демонстрировали фенотип, соответствующий дефекту секреции инсулина с более ранним началом, меньшей массой тела более низким уровнем инсулина [23].
Описанная мутация расположена в трансактивационном домене белка HNF1A, высококонсервативного донорного сайта сплайсинга в аминокислотной позиции 391 и входит во все 3 описанные ранее изоформы белка. Но, судя по всему, патогенный эффект этой мутации связан не с изменением структуры белка, а с изменением набора кодируемых этим геном мРНК. Определяющая ее rs137853240 находится в 3 нуклеотиде от 3’ границы экзона 4 этого гена. Функциональные исследования на клеточных линиях показали, что rs137853240 приводит к образованию двух аномальных укороченных транскриптов и существенному снижению количества полноразмерного транскрипта гена HNF1A. Существует гипотеза, что такой дисбаланс может приводить к снижению общих уровней транскрипции и быть причиной возникновения СД [27]. Принимая во внимание описанные фенотипические особенности лиц-носителей варианта Gly319Ser, мы предполагаем, что этот вариант ассоциирован с развитием MODY.
В настоящем исследовании продемонстрирован пример успешной диагностики СД с нетипичным течением – MODY-HNF1A – путем объединения клинических критериев и метода высокотехнологического секвенирования ДНК. Методы секвенирования нового поколения позволяют более эффективно и экономически выгодно диагностировать типы MODY [28]. Применение метода высокотехнологичного секвенирования также перспективно при оценке возможных причин развития фенотипа MODY в случае отсутствия причинных мутаций в традиционных MODY-ассоциированных генах для обнаружения потенциально патогенных мутаций в других генах.
ЗАКЛЮЧЕНИЕ
Представленный случай показывает перспективность молекулярно-генетического исследования пациентов с семейными формами СД, в частности, связанного с анализом гена HNF1A. Персонализированный подход к диагностике и лечению особенно важен при выявлении неклассического течения СД у лиц молодого возраста.
